微滴式数字PCR技术在基因编辑中的应用
—日读一帖,昆明友宁团队伴你科研路
【科研热点】让你时间比别人花的少、知道的比他早!
【热点促销】让你用最少的钱,了解最新的促销信息!
【实验技能】这么棒快告诉你老板
ddPCR™与编辑效率的评估
基因编辑技术精确靶向修饰基因组的能力,包括功能性敲除内源基因或等位基因,靶向诱导或者纠正特定突变及其他多态性,使其在分子生物学领域有着巨大的应用潜力,正在改变分子生物学研究的思路和方法。TALENs,特别是CRISPR-Cas9系统 使基因编辑在众多实验中成为标准技术手段。在基础研究领域,基因编辑可用于探究基因功能或分析多突变和SNP的影响。除了能在正常细胞中建立精确的疾病模型,基因编辑还可以作为基因治疗工具,用于治疗各种疾病。
序列特异的核酸酶切割基因组产生DSB,激活细胞的DNA修复机制,包括NHEJ和HDR途径。错误倾向的NHEJ可导致短序列的插入或删除,可用来敲除基因功能;对于大多数应用,HDR更为有用,因为HDR利用同源重组和同源供体DNA,能精确的基因修复,而NHEJ会导致不可预知的损伤(序列未知的插入或删除)。实际运用中,HDR编辑的细胞群体往往含有NHEJ导致的等位基因,且NHEJ和HDR之间的平衡难以控制,这是因为缺乏快速而灵敏方法对NHEJ和HDR编辑结果进行准确定量。
对于很多基因编辑应用来说,稳定检测评估基因编辑诱导的突变对于项目的推进是非常重要的。不仅如此,高灵敏度的,通用的,可用于任何靶标的突变定量检测手段也有很大的需求。基因编辑实验通常只能在细胞群体中产生很低的突变频率。因此对于突变频率(编辑效率)的精确定量能力对于优化基因编辑方案以及下游实验流程来说是极端重要的。比如,帮助判断需要筛查多少个单细胞克隆以检出所需的突变。
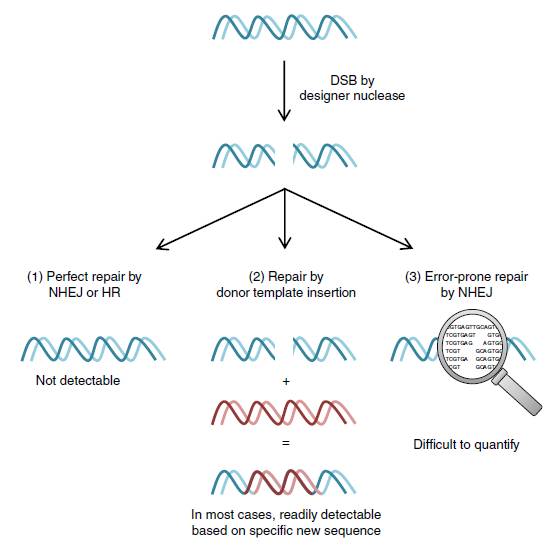
图1. DSB细胞修复结果:(1)通过NHEJ或HDR,用姐妹染色单体完全修复,无法检测;(2)使用同源供体模板,导入特定突变;(3)基于NHEJ,可产生插入/删除突变。GEF-dPCR用以检测NHEJ介导的突变频率
目前常用的基因编辑突变检测方法包括二代测(NGS)、高分辨率熔解曲线(HRM)、T7E1核酸酶切测试、克隆测序等。NGS技术是检测核酸酶诱导未知突变的金标,但是由于需要昂贵的设备和生物信息学支持,耗时较长,费用偏高,因此常常不太可行。其他各种用于核酸酶诱导突变的筛查手段中使用最广的一类方法利用所谓的“错配核酸酶”T7E1,可以识别并酶切含有错配碱基对的异源双链DNA扩增产物。然而这类方法有明显的缺点。
首先,需要大量的样本以制备至少200ng(500ng为最佳)的包含靶标位点的纯扩增产物。这就制约了实验流程的速度,因为流式分选或克隆挑选富集的编辑细胞必须经过充分的培养增殖才能放大数量(即便在获得单细胞克隆之后,仍然需要再进行细胞培养)。
其次,灵敏度明显不够(>1%)。酶切后的片段在靶分子扩增总产物中占比低,在电泳胶上难以从背景干扰中区分出来。靶标序列如果不能有效扩增则难以产生明亮的条带。事实上,由于嵌入式DNA染色的特性,每个酶切片段将损失相对于原始条带至少50%的信号。
再次,这类方法对于单细胞克隆的筛查效果不尽人意。对于一个典型的二倍体靶位点,若两个等位基因都被NHEJ成功编辑,将产生不同的插入/缺失突变,这样的细胞克隆无法与同时含有一个突变等位基因和野生型等位基因的克隆区分开,因为这两种样本都将含有50%:50%混合的不同等位基因。这两种情况下,一半双链DNA将处于双链杂合状态,而另一半将形成纯和双链DNA(25% allele 1和25% allele 2)因而不会被酶解。这样的情况只能通过更多的测序和/或更多克隆筛选来解决,但无疑会导致更多科研资源的投入。
最后,这类方法通常需要制备至少400bp的扩增产物,以保证酶切片段有足够的长度,从而在能凝胶上显示。但是这么长的片段无疑会增加扩增产物包含某种在样本或细胞系中处于杂合状态的多态性位点。扩增产物上任何位置的内源杂合SNP位点或突变型都会被T7E1核酸酶识别,从而导致未被编辑细胞也被酶切,出现假阳性信号。这对于肿瘤细胞系以及突变频率极高,位点未知且动态变化的样本来说是尤为严重的问题。总之,基因编辑效率的定量评估依然是一个瓶颈问题,一定程度上影响了基因编辑技术的应用。

图2. 微滴式数字PCR工作流程
为了解决基因编辑实验中突变定量检测问题,Bio-Rad推出了基于微滴式数字 PCR(ddPCR)的检测方法,为基因编辑效率评估提供了新的方法和思路。被称为第三代PCR技术的ddPCR是一种全新的核酸分子定量检测技术,它通过微滴化过程,将核酸模板随机分装进20000个油包水微滴中,每个微滴都是一个独立的PCR反应器。PCR扩增后只有含有靶标序列的微滴呈阳性,不含靶标序列的微滴呈阴性。通过阳性微滴的比例和泊松分布概率函数,即可得到样本的拷贝数浓度(图2)。与定量PCR相比,其结果的精确度、准确性和灵敏度更佳;定量的结果不再依赖于标准曲线,直接给出靶序列的起始拷贝数浓度,实现真正意义上的绝对定量。
2016年2月的Nature Protocol杂志(IF9.646)报道了德国科学家基于ddPCR建立的基因编辑效率评价新方法(gene-editing frequency digital PCRPCRPCR, GEF-dPCR)。该方法使用了伯乐公司(Bio-Rad)称为“Drop-off”的策略,以双重dtudPCR扩增方式,可同时检测样本中的野生型区域和编辑区域,解决了NHEJ诱导的InDel快速检测的难题。GEF-dPCR体系建立后,研究人员在1天内即可完成对基因编辑样本的编辑率评估(图2)。相比常规的错配核酸酶切法,ddPCR更为准确、灵敏度也更加灵活,可用于任何指定的靶标。因此可以用作定量比较不同基因编辑平台、方法和方案的手段。
Drop-off策略的思路是(图3):针对靶标基因设计两条不同的水解探针,以不同的荧光素标记(FAM和HEX);其中一条探针结合在扩增片段内的非基因编辑靶区域,作为“reference”探针,另外一条结合NHEJ编辑区域,作为“NHEJ/Drop-off”探针。在ddPCR扩增时,未能编辑的野生型模板由于reference探针和NHEJ/Drop-off探针同时水解,同时产生FAM和HEX双色荧光,在ddPCR 2-D视图中,显示为FAM和HEX双阳性的微滴(含有野生型序列);成功编辑的序列,由于靶标区域发生InDel,NHEJ/Drop-off探针无法结合,只有reference探针水解,产生FAM荧光,在ddPCR 2-D视图中显示为FAM阳性的微滴(含有编辑后序列)。
QX200TM ddPCR不仅具有更高的灵敏度,能绝对定量的特点,还易于调整方案以适用于任何需编辑靶基因,可分别或同时检测NHEJ和HDR诱导突变;同时ddPCR方法简便易行,通量大,检测速度快,体系建立后1天内即可完成大量样本的编辑效率评估,还可用于编辑细胞的快速富集培养,因此QX200 ddPCR很有希望成为基因编辑效率评估的首选方法,推动基因编辑技术应用的发展。
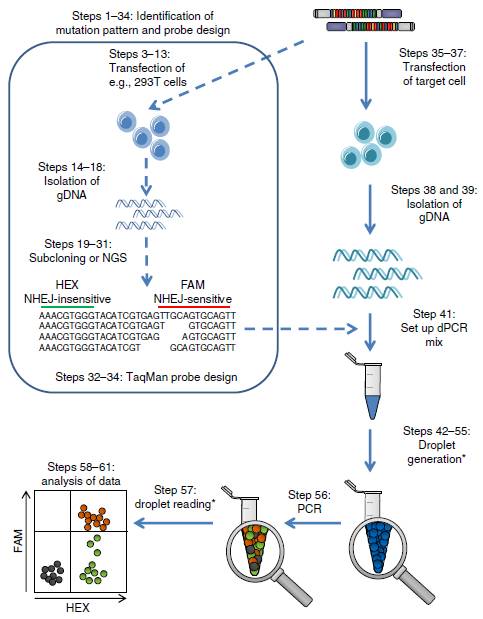
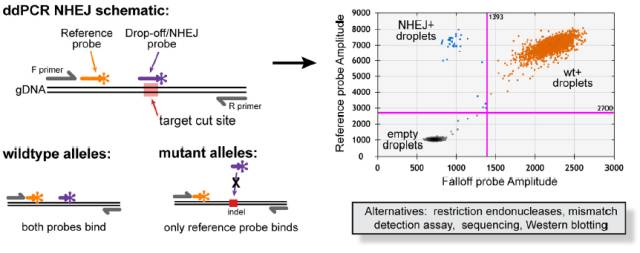
图3. Drop-off探针设计和检测原理
推荐文献
1. Lander N, et al. (2015) CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. mBio 6(4):e01012-15.
2. Sedlak, et al. (2106) Digital detection of endonuclease mediated gene disruption in the HIV provirus Sci Rep. Feb 2;6:20064.
3. Mock, et al. (2016) Digital PCR to assess gene-editing frequencies (GEF-dPCR) mediated by designer nucleases. Nat Protoc. Mar;11(3):598-615.
4. Mock et al. (2015) mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Res. May 11.
Miyaoka et al. (2014) Isolation of single-base genome edited human iPS cells without antibiotic selection. Nat Methods. Mar;11(3):291-3.