火山图—基因的差异表达
 什么是火山图
什么是火山图
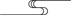
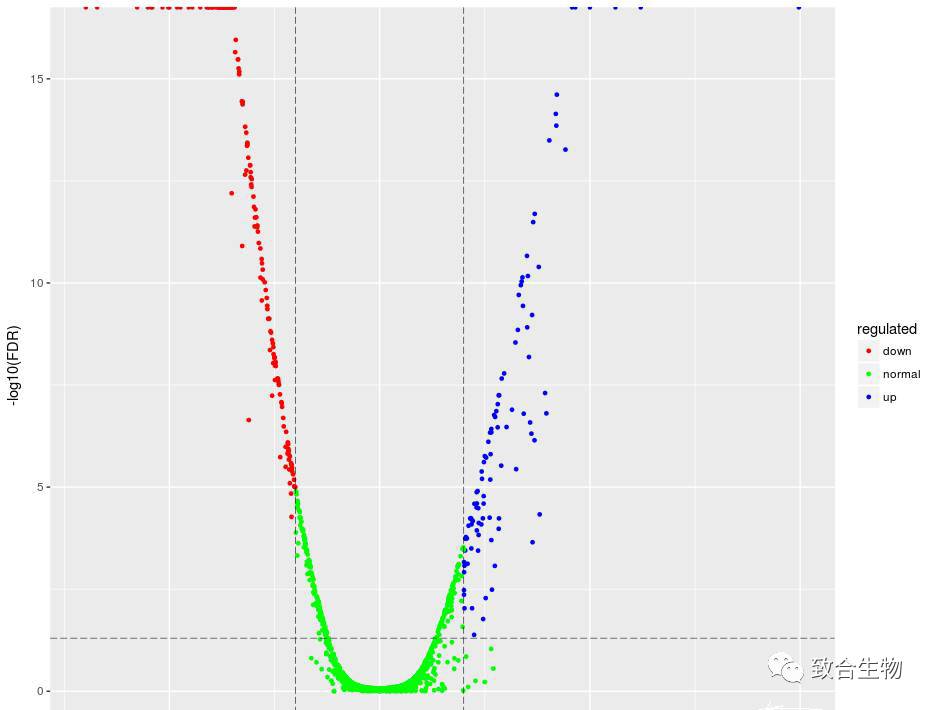
火山(Volcano Plot)图在一张图中显示了两个重要的指标(Foldchange/pvalue),可以非常直观且合理地筛选出在两样本间发生差异表达的基因。检验分析出两样本间显著差异表达的基因后,以log2(fold change)为横坐标,以T检验显著性检验P值的负对数-log10(pvalue)为纵坐标,即可得火山图(Volcano Plot)。
火山图怎么制作呢
火山图(Volcano Plot)可以直观地展现所有基因的FDR与Fold Change之间的关系,以便快速查看基因在两组样品间的表达水平差异程度及其统计学显著性。
两个重要概念:FDR和FC
FDR:在差异表达分析过程中,采用Benjamini-Hochberg方法对原有假设检验得到的显著性p值(p-value)进行校正,并最终采用校正后的p值,即FDR(False Discovery Rate)。
FC(Fold Change):两样品(组)间表达量的比值。
我们简单演示一下
#载入包
library(ggplot2)
#导入数据
df<-read.delim(file = "test.xls",header = T,sep = "\t",check.names = F,row.names = 1,stringsAsFactors = F)
#简单查看
head(df,n=10)
针对数据进行处理,在这里根据两(组)样品之间表达水平的相对高低,差异表达基因可分为上调基因(upregulated Gene)和下调基因(down)。上调和下调是相对的,由所给A和B的顺序决定,若更换A和B的顺序会上调基因和下调基因反过来,但对结果没有影响。
我们根据FDR<=0.05和|FC|>=2作为筛选标准!
for (i in 1:nrow(df) ) {
if ( as.double(df[i,ncol(df)-1]) >= 2 && as.double(df[i,ncol(df)-2]) <= 0.05 ) {
df[i,ncol(df)] <- "up"
} else if ( as.double(df[i,ncol(df)-1]) <= -2 && as.double(df[i,ncol(df)-2]) <= 0.05 ) {
df[i,ncol(df)] <- "down"
} else {
df[i,ncol(df)] <- "normal"
}
}
然后呢,就是利用ggplot2进行绘制了……
p <- ggplot(df, aes(x=log2FC, y=-log10(FDR), colour=regulated) ) + geom_point(size=1)
p <- p + scale_color_manual(values = c('red',"green","blue"))
p <- p + geom_vline(xintercept=c(-2,2), linetype="longdash", size=0.2)
p <- p + geom_hline(yintercept=c(-log10(0.05)), linetype="longdash", size=0.2)
p
差异表达火山图中的每一个点表示一个基因,横坐标表示某一个基因在两样品中表达量差异倍数的对数值,其绝对值越大,说明表达量在两样品间的表达量倍数差异越大;纵坐标表示FDR的负对数值,其值越大,表明差异表达越显著,筛选得到的差异表达基因越可靠。图中红色和蓝色的点代表有显著性表达差异的基因,红色代表基因表达量下调,蓝色代表基因表达量上调,绿色的点代表无显著性表达差异的基因。
拓展
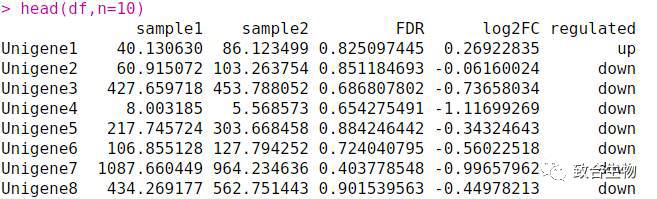
MA图可以直观地查看两组样品中基因的表达丰度和差异倍数的整体分布!
#统计上调基因和下调基因的数目
normal<-paste0("normal:",sum(df[,"regulated"] == "normal"))
up<-paste0("up:",sum(df[,"regulated"] == "up"))
down<-paste0("down:",sum(df[,"regulated"] == "down"))
sig <- c(normal,up,down)
#绘制MA图
v<-c("sample1","sample2")
data=data.frame(x=log2(rowMeans(df[,v])), y=df[,c("log2FC")],lab=factor(df[,"regulated"],levels=unique(df[,"regulated"])))
p=ggplot(data,mapping=aes(x=log2(rowMeans(df[,v])), y=df[,c("log2FC")],color=lab))
p=p+geom_point(size=1)
p=p+xlab("log2(FPKM)")+ylab("log2(FC)")+ggtitle("MA plot")+theme(plot.title=element_text(face="bold",size=14))
p=p+theme_classic()
p=p+scale_color_manual(name="regulated",values=c("red","green","blue"),labels=sig)
p
这里我们提供另外一种绘制火山图的方法:
library(ggplot2)
data=read.table(file="deg-result.txt",header=T,row.names=1)
threshold<-as.factor((data
logFC< -1.5)& data$P.Value<0.05)
ggplot(data,aes(x=logFC,y=-log10(P.Value),colour=threshold))+xlab("log2fold change")+ylab("-log10p-value")+geom_point()
关于p值
p-value或q-value是统计学检验变量,代表差异显著性,一般p-value或q-value小于0.05代表具有显著性差异,但可根据具体情况适当调整。
因为p-value或q-value衡量地是某个基因假阳性的概率,如果p-value或q-value越低,那么挑选该基因出现假阳性的概率就越低,可验证性就越高。
那p-value、q-value同时存在时看哪个呢?
SAM法只有q-value。当两者同时存在时,可根据具体情况具体分析。
差异筛选是一个典型的多重假设检验过程,对于多重假设检验,单次检验中差异显著基因的假阳性率(p-value较小)可能会较大,而q-value和FDR值较常见的BH校正方法得到的FDR值而言,改进了其对假阳性估计的保守性。
即q-value相比于p-value更加严格,当差异基因结果较少时,可以退而求其次看p-value。
Fold ChangeFold Change表示实验组比上对照组的差异表达倍数,一般表达相差2倍以上是有意义的,放宽要求1.5倍或者1.2倍也可以接受。看表达倍数的同时还需结合基因表达丰度,信号值太低的基因会在后续的验证实验中检测不到。