用deFuse来对转录组数据找融合基因
首先提醒一下,该工具需要下载 108 G 的数据库文件才能运行,而且仅仅是针对hg19这一个参考基因组。但是因为其发表的非常早,即使不好用,也仍然是目前最主流的转录组数据找融合基因工具之一。
该工具发表于 2011,文章是deFuse: An Algorithm for Gene Fusion Discovery in Tumor RNA-Seq Data 软件,其readme写的非常简陋:https://sourceforge.net/p/defuse/wiki/DeFuse/ ,不过其bitbucket上面稍微详细一点; https://bitbucket.org/dranew/defuse
但是可以用conda来安装,不过其依赖比较多,如果conda没有配置好,估计好几天才能安装成功。
conda install -c dranew defuse
因为在海外,所以几分钟就OK拉,或者下载源代码自己安装咯
mkdir -p ~/biosoft/defuse
cd ~/biosoft/defuse
wget https://sourceforge.net/projects/defuse/files/defuse/0.6/defuse-0.6.2.tar.gz
## 依赖非常多的软件,自己安装太费劲
tar zxvf defuse-0.6.2.tar.gz
head ~/biosoft/defuse/defuse-0.6.2/scripts/config.txt ## 这个配置文件需要修改的东西太多了。
运行该软件
直接从fastq数据开始,它自己承包了比对这个任务,所以需要非常复杂的参考基因组及注释文件数据库构建。虽然它只有一个代码即可,但是耗费一整天的时间下载数据库。
运行也非常简单,示例代码如下:
defuse_create_ref.pl -d dataset_directory -c myconfig.txt ## 超过12个小时
run_defuse.pl -d dataset_directory -1 reads1.fq -2 reads2.fq -o output_dir
上面的代码会使用 myconfig.txt文件,在该软件的源代码压缩包里面有示例文件 ~/biosoft/defuse/defuse-0.6.2/scripts/config.txt ,需要修改适应自己的系统,主要是修改 source_directory 和 dataset_directory ,详细说明如下:
#
# Configuration file for defuse
#
# At a minimum, replace all values enclosed by []
#
# For example:
# source_directory = /path/to/defuse
#
ensembl_version = 69
ensembl_genome_version = GRCh37
ucsc_genome_version = hg19
# Directory where the defuse code was unpacked
source_directory = [Where you unpacked the defuse code]
# Directory where you want your dataset
dataset_directory = [Where you intend to store the reference dataset]
# Input genome and gene models
gene_models = $(dataset_directory)/Homo_sapiens.$(ensembl_genome_version).$(ensembl_version).gtf
genome_fasta = $(dataset_directory)/Homo_sapiens.$(ensembl_genome_version).$(ensembl_version).dna.chromosomes.fa
太麻烦,所有的中括号里面的东西都需要修改,该软件默认调用 gmap这个软件来做转录组数据的比对,所以还需要提供gmap的转录组索引文件。
但是因为是用conda进行安装的,所以一切都被安排好了,只需要找到自己想存放数据的地址,然后运行即可。
mkdir -p ~/biosoft/defuse/database/
nohup defuse_create_ref.pl -d ~/biosoft/defuse/database/ &
## 还是取决于网速,需要下载 108G 的文件, 仅仅是针对hg19这个参考基因组
## 84G /home/jianmingzeng/biosoft/defuse/database/gmap
## 108G /home/jianmingzeng/biosoft/defuse/database/
## 自己检查 ~/biosoft/defuse/database/ 文件夹的下载情况咯。
### 该脚本会自动调用conda为defuse配置好的config.txt
## 如果是其它物种,可以修改 ~/miniconda3/pkgs/defuse-0.8.1-r3.3.2_0/opt/defuse/scripts/config.txt 文件
cd data/project/fusion/defuse/
fq1='/home/jianmingzeng/data/project/fusion/clean/clean.1.fq'
fq2='/home/jianmingzeng/data/project/fusion/clean/clean.2.fq'
sample='test'
defuse_run.pl -d ~/biosoft/defuse/database/ -p 5 -1 $fq1 -2 $fq2 -o $sample
这个软件很难用,居然不支持gz格式压缩好的fastq测序数据!
软件运行过程也很无语,居然会把所有的fastq文件拷贝到运行目录,并且拆分成小文件。都是在耗费硬盘空间呀,完全不建议用云服务器运行,毕竟云服务器最贵的就是硬盘存储了。
虽然我写了教程,但是真心不建议继续使用这个软件!
结果文件非常复杂,如果想完全看懂,建议还是自己看英文介绍吧
Identification
cluster_id : random identifier assigned to each prediction
library_name : library name given on the command line of deFuse
gene1 : ensembl id of gene 1
gene2 : ensembl id of gene 2
transcript1 : transcript id of gene 1, or 'NA' for non-exonic fusions
transcript2 : transcript id of gene 2, or 'NA' for non-exonic fusions
gene_name1 : name of gene 1
gene_name2 : name of gene 2
Evidence
break_predict : breakpoint prediction method, denovo or splitr, that is considered most reliable
concordant_ratio : proportion of spanning reads considered concordant by blat
denovomincount : minimum kmer count across denovo assembled sequence
denovo_sequence : fusion sequence predicted by debruijn based denovo sequence assembly
denovospanpvalue : p-value, lower values are evidence the prediction is a false positive
genealignstrand1 : alignment strand for spanning read alignments to gene 1
genealignstrand2 : alignment strand for spanning read alignments to gene 2
minmapcount : minimum of the number of genomic mappings for each spanning read
maxmapcount : maximum of the number of genomic mappings for each spanning read
meanmapcount : average of the number of genomic mappings for each spanning read
nummultimap : number of spanning reads that map to more than one genomic location
span_count : number of spanning reads supporting the fusion
span_coverage1 : coverage of spanning reads aligned to gene 1 as a proportion of expected coverage
span_coverage2 : coverage of spanning reads aligned to gene 2 as a proportion of expected coverage
spancoveragemin : minimum of spancoverage1 and spancoverage2
spancoveragemax : maximum of spancoverage1 and spancoverage2
splitr_count : number of split reads supporting the prediction
splitrminpvalue : p-value, lower values are evidence the prediction is a false positive
splitrpospvalue : p-value, lower values are evidence the prediction is a false positive
splitr_sequence : fusion sequence predicted by split reads
splitrspanpvalue : p-value, lower values are evidence the prediction is a false positive
Annotation
adjacent : fusion between adjacent genes
altsplice : fusion likely the product of alternative splicing between adjacent genes
breakadjentropy1 : di-nucleotide entropy of the 40 nucleotides adjacent to the fusion splice in gene 1
breakadjentropy2 : di-nucleotide entropy of the 40 nucleotides adjacent to the fusion splice in gene 2
breakadjentropymin : minimum of breakadjentropy1 and breakadj_entropy2
breakpoint_homology : number of nucleotides at the fusion splice that align equally well to gene 1 or gene 2
breakseqsestislandspercident : maximum percent identity of fusion sequence alignments to est islands
cdnabreakseqspercident : maximum percent identity of fusion sequence alignments to cdna
deletion : fusion produced by a genomic deletion
estbreakseqspercident : maximum percent identity of fusion sequence alignments to est
eversion : fusion produced by a genomic eversion
exonboundaries : fusion splice at exon boundaries
expression1 : expression of gene 1 as number of concordant pairs aligned to exons
expression2 : expression of gene 2 as number of concordant pairs aligned to exons
gene_chromosome1 : chromosome of gene 1
gene_chromosome2 : chromosome of gene 2
gene_end1 : end position for gene 1
gene_end2 : end position for gene 2
gene_location1 : location of breakpoint in gene 1
gene_location2 : location of breakpoint in gene 2
gene_start1 : start of gene 1
gene_start2 : start of gene 2
gene_strand1 : strand of gene 1
gene_strand2 : strand of gene 2
genomebreakseqspercident : maximum percent identity of fusion sequence alignments to genome
genomicbreakpos1 : genomic position in gene 1 of fusion splice / breakpoint
genomicbreakpos2 : genomic position in gene 2 of fusion splice / breakpoint
genomic_strand1 : genomic strand in gene 1 of fusion splice / breakpoint, retained sequence upstream on this strand, breakpoint is downstream
genomic_strand2 : genomic strand in gene 2 of fusion splice / breakpoint, retained sequence upstream on this strand, breakpoint is downstream
genomic_starts1 : comma separated list of starts of fusion sequence alignment in gene 1
genomic_starts2 : comma separated list of starts of fusion sequence alignment in gene 2
genomic_ends1 : comma separated list of ends of fusion sequence alignment in gene 1
genomic_ends2 : comma separated list of ends of fusion sequence alignment in gene 2
interchromosomal : fusion produced by an interchromosomal translocation
interrupted_index1 : ratio of coverage before and after the fusion splice / breakpoint in gene 1
interrupted_index2 : ratio of coverage before and after the fusion splice / breakpoint in gene 2
inversion : fusion produced by genomic inversion
orf : fusion combines genes in a way that preserves a reading frame
probability : probability produced by classification using adaboost and example positives/negatives (only given in results.classified.tsv)
read_through : fusion involving adjacent potentially resulting from co-transcription rather than genome rearrangement
repeat_proportion1 : proportion of the spanning reads in gene 1 that span a repeat region
repeat_proportion2 : proportion of the spanning reads in gene 2 that span a repeat region
maxrepeatproportion : max of repeatproportion1 and repeatproportion2
splice_score : number of nucleotides similar to GTAG at fusion splice
numsplicevariants : number of potential splice variants for this gene pair
splicing_index1 : number of concordant pairs in gene 1 spanning the fusion splice / breakpoint, divided by number of spanning reads supporting the fusion with gene 2
splicing_index2 : number of concordant pairs in gene 2 spanning the fusion splice / breakpoint, divided by number of spanning reads supporting the fusion with gene 1
融合基因背景知识
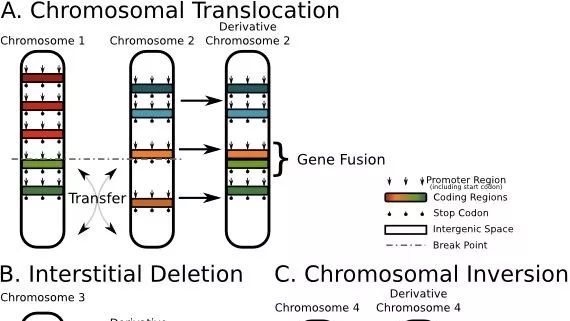
融合基因(Fusion gene)是指两个基因的全部或一部分的序列相互融合为一个新的基因的过程。其有可能是染色体易位、中间缺失或染色体倒置所致的结果。
异常的融合基因可以引起恶性血液疾病以及肿瘤。例如典型的EML4-ALK BCR-ABL融合基因可以导致白血病,此外还有在前列腺癌症里面经常被发现的TMPRSS2-ERG,在非小细胞肺癌里面经常发现的EML4-ALK,VTI1A-TCF7L2 (直肠癌)。
目前已经很多工具,基于高通量测序数据来对检测融合基因。例如:soapfuse,FusionSeq , deFuse, TopHat-Fusion, Fusion- Hunter, SnowShoes-FTD, chimerascan ,FusionMap和STAMP。
参考:https://bioinformaticsdotca.github.io/BiCG2017module4genefusionsinstallation
最后说一句,这个工具,能不用,就不用吧,说多了都是累。