罕见病基因治疗的研究进展
来源:《国际药学研究杂志》微信平台

【摘要】罕见病因发病率低,治疗药物的市场需求低,导致其治疗药物的研发成本过高,相关治疗策略进展缓慢。近年来,随着分子生物学技术的发展和精准医疗概念的提出,基因治疗技术在遗传性罕见病的研究中取得了重大进展,其研究成果在罕见病的临床诊断、药物研发和治疗中发挥着重要作用,为彻底治愈疾病提供了可能。本文综述了基因治疗的主要原理、策略和在罕见病中的潜在应用,重点介绍了基因编辑技术在罕见病治疗中的优势,并总结了近年来基因治疗相关的临床试验,为基因治疗在罕见病精准医疗领域的研究和应用提供参考。
罕见病又名“孤儿病”,是指发病率极低的少见疾病。部分可危及生命。在罕见病的患者中,近半数在出生时或儿童时期即可发病,但是目前只有不到5%的罕见病种获得有效的治疗。
罕见病的发病机制比较复杂,约有80%的罕见病是由遗传缺陷引起,依据发病机制的差异,可采取不同的治疗策略。罕见病的治疗主要有饮食治疗、药物治疗、手术治疗、骨髓移植治疗和基因治疗等多种方法。在基因水平对各种罕见疾病进行精准治疗,是目前医学研究的重点,也是未来相关新药研究开发的重要方向之一。本文主要介绍基因治疗在罕见病研究和治疗中的潜在应用。
1 基因治疗的策略
基因治疗,是指对基因进行纠正或置换突变致病基因的一种治疗方法。将目的基因导入靶细胞内,与宿主细胞染色体整合成宿主遗传物质的一部分,或不与染色体整合而位于染色体外,均能在细胞中得到表达,从而达到治疗疾病的作用。新兴的基因编辑技术的兴起,增加和扩宽了基因治疗在疾病治疗中的应用。
基因治疗策略的选择对于疾病治疗的成功至关重要。根据不同的病因和病理变化可采用不同的治疗策略。
1. 1 基因置换或基因增补
用正常的基因对基因组中突变的基因在原位进行置换或矫正,这种治疗方式称为基因置换。该法的主要原理是采用基因的定点重组技术,将某个具有正常功能的基因或表达元件,通过载体导入到由基因突变而导致功能异常的细胞中,或体内其他存在基因缺陷的部位,从而通过表达完整的蛋白来补充该基因缺失的功能。该法在理论上是最理想的基因治疗策略,缺点在于受到致病基因显隐性的影响,只适用于隐性致病基因,例如多种血友病相关突变基因的治疗[1-2]。而对显性的致病基因则无法通过功能补充来掩盖基因突变带来的影响。
1. 2 基因可调控表达
一些疾病的基因治疗不仅需要基因的正确表达,还需要实现基因的可调控表达。比较常用的小分子药物,包括四环素调控、西罗莫司(雷帕霉素)调控等,miRNA 的转录后调控也被应用到基因的时序表达设计中。近年来,光调控、磁调控的基因表达研究日益增多。安全可靠的基因可调控表达系统无疑是基因治疗技术中的重要环节,也是基因治疗愈发精准的重要体现。
1. 3 基因编辑技术
基因编辑技术可在基因组水平上对DNA序列进行精确改造,可对目的基因进行基因敲除,引入特异突变及定点转基因,是真正意义上的精准治疗。利用基因编辑技术可使基因组中突变的基因失活或者纠正突变的基因,将致病基因改造为正常基因,实现治疗疾病的目的。目前常用的基于人工核酸酶的基因编辑技术,可在靶标位点对基因实现精确的编辑或敲除,进而实现基因治疗疾病的目的。
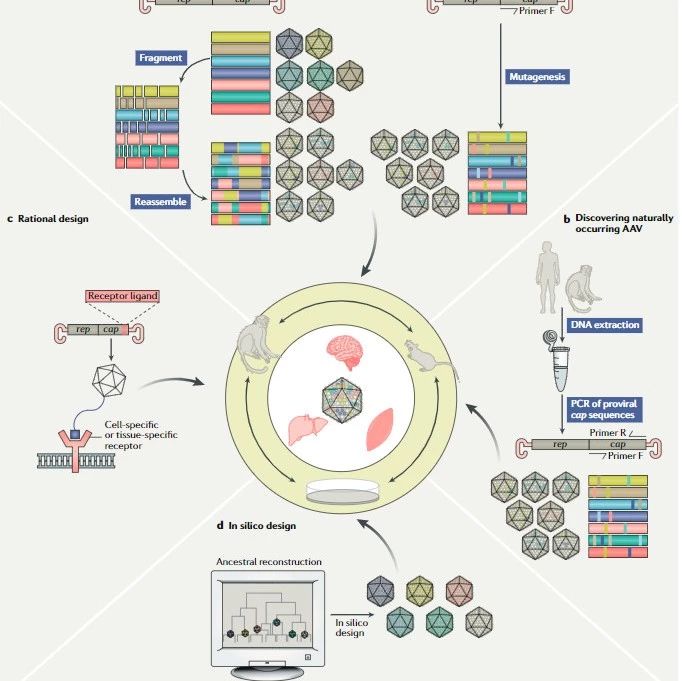
传统的基因组编辑技术是利用胚胎干细胞(embryonic stem cell,ES)与同源重组技术,对基因组进行定点修饰。该技术依赖于ES细胞的获得及重组效率等因素,具有效率低、耗时长等缺点。而基于人工核酸酶的基因组编辑技术,则具有效率高、特异性强等优点[3],可以定点改造基因组。目前常用的基于人工核酸酶的3种基因编辑技术是:锌指核酸酶(zinc finger nucleses,ZFN)、类转录激活因子核酸酶(transcription activator-like effector nucleases,TALEN)和规律成簇的间隔短回文重复序列/CRISPR相关蛋白系统(clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9,CRISPR/Cas9)。这三类人工核酸酶都由DNA识别区域和DNA切割区域组成,可在预定的基因组位置切断DNA,切断的DNA在被细胞内的DNA修复系统修复过程中会产生随机突变或定制的序列改变,从而实现定点改造基因。其中ZFN和TALEN的DNA识别区域通过氨基酸识别碱基,然后利用限制性内切酶FokI对DNA进行切割[4]。而CRISPR/Cas9系统通过碱基互补配对原则由CRISPR向导RNA识别DNA序列,之后由与向导RNA结合的Cas9核酸酶切割DNA[5]。CRISPR/Cas9系统较其他两种技术具有明显优势,基因定位更简单易用,可更高效地同时识别多个基因序列。
2 基因治疗在罕见病治疗研究中的应用
自2006年以来,基因治疗在医学研究领域取得很多进展,安全性和有效性有了很大提升。目前已经在多种疾病治疗中取得了成果,如囊性纤维化[6]、血友病[7]、恶性肿瘤[8]、神经变性疾病[9]、传染病[10]和严重的病毒感染(如艾滋病)[11]等。随着CRISPR技术的蓬勃兴起,通过基因编辑技术有望达到根治疾病的目的,这种新兴的基因治疗方式是目前研究的一大热点[12]。
2. 1 基因编辑技术在罕见病研究中的应用
基于人工核酸酶的基因编辑技术可在靶标位点对基因实现精确的编辑或敲除,进而实现基因治疗疾病的目的。CRISPR/Cas9系统由于其使用方法的简单易行,在人类疾病基因治疗中具有良好的应用前景,是目前应用最广发展最快的基因编辑系统。基因编辑技术可用于建立罕见病动物模型,促进罕见病发病机制研究和药物研发。Chen等[13]使用CRISPR/Cas9靶向猕猴的肌营养不良蛋白(dystrophin)编码基因,使其发生可导致杜氏(Duchenne)肌营养不良症的突变。靶向效率检测结果表明,CRISPR/Cas9靶向技术可导致猕猴肌肉中87%的肌营养不良蛋白基因发生嵌合突变,从而成功构建了肌营养不良的疾病模型。
CRISPR/Cas9系统可能为人类遗传疾病的治疗提供新的策略。Guan等[14]发现,CRISPR/Cas9系统可以修复血友病小鼠肝细胞0.56%的F9突变基因,并足以恢复止血功能。2013年初,丛乐等[4]首次利用CRISPR/Cas9系统,在人类与小鼠细胞系中实现了基因敲除。如Mali等[15]利用改造的CRISPR/Cas9系统在人类不同的基因靶点形成单链或双链切口,然后激活细胞的DNA修复机制,高效介导定点突变和外源基因的定点插入。Xie等[16]将地中海贫血症患者的皮肤细胞诱导分化为诱导多功能干细胞(inducible pluripotent stem cell, iPSC),利用CRISPR/Cas9系统修复HBB基因,修复后的iPSC诱导分化出的红细胞可以稳定表达正常的β珠蛋白。Canver等[17]利用CRISPR/Cas9系统可在人类造血干细胞中敲除BCL11A基因表达的增强子基因,从而使HbF基因的表达量升高,进而缓解镰状细胞贫血患者的贫血症状[18]。2014年,Nature和Science杂志分别报道了运用CRISPR/Cas9技术治疗FAH基因突变的酪氨酸血症和肌营养不良蛋白基因突变的杜氏肌营养不良症[19]。
CRISPR/Cas9系统在基因治疗遗传疾病等领域具有良好应用前景,但是也存在脱靶效应等弊端。脱靶效应可能会导致基因组中其他编码蛋白的基因序列改变、激活癌基因等不良后果,给临床应用带来风险。另外,如何有效将Cas9基因组编辑系统准确运送到机体特定细胞和组织,实现精确导向的基因治疗,也是当今基因编辑技术在临床应用的一大难题。基因药物递送系统的发展,对基因编辑技术的临床应用发挥关键作用。
2. 2 基因治疗药物递送系统的发展
基因治疗药物输送系统在基因治疗中起到至关重要的作用,它有效增强了基因治疗药物的稳定性和靶向性,达到了更好的治疗效果。对于很多罕见病,如肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)、脊髓性肌萎缩(spinal muscular atrophy,SMA),目前尚无药物可以进行有效的治疗。
SMA是儿童遗传性疾病,是由于运动神经元退化和进行性肌无力而导致婴儿死亡的主要遗传性疾病[20-21]。基于基因治疗的一种治疗方法是利用单链反义剪接转换寡聚核苷酸(splicing-switching oligonucleotides,SSO)通过空间阻断pre-mRNA的剪接调节来获得运动神经元生存基因(survival motor neuron gene,SMN)2包含第7个外显子的pre-mRNA[22]。尽管SSO治疗在当前发展迅速,也是SMA最有希望的治疗方法之一。但因SSO不能穿过血脑屏障,并且必须通过重复的鞘内注射,近1/3的患者需额外承受腰椎穿刺相关的风险[23]。近期研究报道了一种二亚磷酰胺低聚物(phosphorodiamidate oligomer,PMO)内化肽(PMO internalizing peptide,Pip)递送技术,可改善中性带电SSO的递送[24]。Pip肽是共价缀合的,能够将SSO递送到多种成人组织中,包括肝、肾、骨骼肌、隔膜和心脏。研究发现,高活性肽Pip6a直接结合到吗啉基的PMO上,可以增强包括脑和脊髓在内的全身SMN的表达、明显延长严重SMA小鼠的生存期[25]。这些数据显示通过肽-PMO治疗,在小鼠SMA疾病模型中获得很好的疗效,这种成功的方法也可以扩展到其他退行性疾病的治疗。
2. 3 基因治疗相关的罕见病临床试验
基因疗法治疗失明的首例试验是在2007年,受试对象为10名患有雷伯氏先天性黑朦症(Leber congenital amaurosis, LCA)的志愿者。试验将一种携带RPE65正常基因拷贝的无害病毒注入受试者眼中,治疗后受试者视力得到明显改善。2015年Manzar等[26]发表了用基因疗法治疗LCA患者的成果。回顾性病例对照试验表明,向LCA患者视网膜中注射携带治疗基因的载体,与对照组相比,患者视觉得以持续性改善。向患者眼部注入治疗基因以修复突变已成为先天性和退行性失明最具潜力的治疗方法。CRISPR/Cas9发明人之一张锋博士参与创始的Editas Medicine公司使用基因编辑技术治疗LCA的特定亚型患者,针对由CEP290基因突变引起的该病。CEP290基因较大,无法用病毒装载正常基因的拷贝,而CRISPR/Cas9系统可用腺相关病毒递送,解决这个难题。
2015年Hoban等[27]用基因编辑方法纠正引起镰刀型细胞贫血症的突变基因。这是首次利用基因修正的方法来生成正常的人类红细胞。Bilton等[28]对136名囊性纤维化患者进行了Ⅱ期临床试验,通过植入DNA分子代替突变的囊性纤维化跨膜调节因子相关基因,从而使囊性纤维化患者肺功能得到改善。肺功能轻微损伤者接受基因疗法后肺功能改善达6%以上。
2016年,Hache等[24]利用iPSC和体细胞核移植两项技术,通过收集受试者皮肤细胞,将细胞核植入健康供体细胞的细胞质中,创造出有健康线粒体的胚胎干细胞。通过该线粒体置换技术,为包含线粒体DNA突变的患者构建出了具有健康线粒体的多能干细胞,为通过基因治疗方法治疗线粒体缺陷相关罕见病开启了新方向。
3 结语
本文综述了基因治疗的策略以及在罕见病中的应用,着重介绍了CRISPR/Cas9系统为代表的基因编辑技术的原理和研究进展。近年来基因治疗在LCA、囊性纤维化及线粒体缺陷等疾病的治疗中取得了重大进展,为罕见病患者带来希望。基因编辑技术的临床应用还要依赖于药物投送系统的发展,随着药物投送系统安全性和稳定性的不断提升,基因治疗在罕见病中的应用会不断取得突破。
【参考文献】
[1] Ponder KP, Hemophilia gene therapy: a Holy Grail found [J]. Mol Ther, 2011, 19(3): 427-428.
[2] Matrai J, Chuah MK, VandenDriessche T, Preclinical and clinical progress in hemophilia gene therapy [J]. Curr Opin Hematol, 2010, 17(5): 387-392.
[3] Perez-Pinera P, Chen ZY, Biomedical applications of gene editing [J]. Human Genetics, 2016, 135(9): 967-969.
[4] Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems [J]. Science, 2013, 339(6121): 819-823.
[5] Tasan I, Jain S, Zhao H. Use of genome-editing tools to treat sickle cell disease [J]. Hum Genet, 2016, 135(9): 1011-1028.
[6] Mitomo K, Griesenbach U, Inoue M, et al. Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with Sendai virus envelopes [J]. Mol Ther, 2010, 18(6): 1173-1182.
[7] Walsh CE, Gene therapy progress and prospects: gene therapy for the hemophilias [J]. Gene Ther, 2003, 10(12): 999-1003.
[8] McNeish IA, Bell SJ, Lemoine NR. Gene therapy progress and
prospects: cancer gene therapy using tumour suppressor genes [J]. Gene Ther, 2004, 11(6): 497-503.
[9] Tuszynski MH. Growth-factor gene therapy for neurodegenerative disorders [J]. Lancet Neurol, 2002, 1(1): 51-57.
[10] Bunnell BA, Morgan RA, Gene therapy for infectious diseases [J]. Clini Microbiol Rev, 1998, 11(1): 42.
[11] Morgan RA, Anderson WF. Gene Therapy for AIDS[M]. Springer US. 1991:277-289.
[12] 支大龙, 季维智, 牛昱宇. 后基因组时代的基因编辑技术 [J]. 科学:上海, 2015, 67(6): 28-31.
[13] Xie F, Ye L, Chang JC, et al. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac [J]. Genome Res, 2014, 24(9): 1526-1533.
[14] Guan Y, Ma Y, Li Q, et al, CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse [J]. EMBO Mol Med, 2016, 8(5): 477-488.
[15] Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9 [J]. Science, 2013, 339(6121): 823-826.
[16] Long C, McAnally JR, Shelton JM, et al. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA [J]. Science, 2014, 345(6201): 1184-1188.
[17] Canver MC, Smith EC, Sher F, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis [J]. Nature, 2015, 527(7577): 192-197.
[18] Xu J, Peng C, Sankaran VG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing [J]. Science, 2011, 334(6058): D993-D996.
[19] Chen Y, Zheng Y, Kang Y, et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9 [J]. Hum Mol Genet, 2015, 24(13): 3764-3774.
[20] Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene [J]. Cell, 1995, 80(1): 155-165.
[21] Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) [J]. Hum Mutat, 2000, 15(3): 228-237.
[22] Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1 [J]. Nat Genet, 2002, 30(4): 377-384.
[23] Hammond SM, Wood MJ. Genetic therapies for RNA mis-splicing diseases [J]. Trends Genet, 2011, 27(5): 196-205.
[24] Hache M, Swoboda KJ, Sethna N, et al. Intrathecal injections in children with spinal muscular atrophy: Nusinersen Clinical Trial Experience [J]. J Child Neurol, 2016, 31(7): 899-906.
[25] Hammond SM, Hazell G, Shabanpoor F, et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy [J]. Proceeding Nat Acad Sci USA, 2016, 113(39): 10962-10967
[26] Ashtari M, Zhang H, Cook PA, et al. Plasticity of the human visual system after retinal gene therapy in patients with Leber′s congenital amaurosis [J]. Sci Transl Med, 2015, 7(296): 296-310.
[27] Hoban MD, Cost GJ, Mendel MC, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells [J]. Blood, 2015, 125(17): 2597-2604.
[28] Bilton D, A new chapter in therapy for cystic fibrosis [J]. Lancet Respir Med, 2015, 3(7): 20.
原文刊载:《国际药学研究杂志》2017,44(2)123-126页