基因集富集分析(GSEA)中的排序指标:它们重要吗?
1
摘要
背景
在不同的实验条件下,有许多方法可以用来描述分子途径或基因本体中基因表达变化之间的复杂关系。其中,基因集富集分析(GSEA)似乎是最常用的方法之一(超过10000条引文)。影响最终结果的一个重要参数是基因排序指标的选择。应用默认的排名指标可能会导致糟糕的结果。
方法和结果
本研究采用28个基准数据对16种不同排序指标(包括新方案)进行基因集分析的敏感性和假阳性率进行评价。此外,还测试了所选方法对样本大小的稳健性。采用k-means算法,建立了总体灵敏度、总体误报率和计算负荷最高的四个指标,即适度Welch检验统计量的绝对值(MWT)、最小显著性差(MSD)、信噪比绝对值(S2T)和Baumgartner-Weiss-Schindler(BWS)检验统计量。在误报率估计的情况下,所有选择的排序指标对样本大小都是稳健的(鲁棒性)。在灵敏度方面,稳健的MWT的绝对值和S2T的绝对值给出了稳定的结果,而BWS和MSD则在较大样本量下表现出更好的结果。最后,在matlab程序中并行化并实现了包含所有测试排序指标的基因集富集分析方法,网上可用https://github.com/ZAEDPolSl/MrGSEA。
结论:基因集富集分析中排序指标的选择对通路富集分析的结果有重要影响。在基因集分析中,MWT的绝对值总体灵敏度最高,显着差异最小,总体特异性最好。当非正态分布基因数目较多时,采用BWS检验统计结果较好。此外,还发现了比其他测试指标更富集的通路,这可能会导致新的生物学发现。
关键词:GSEA、排序指标、通路分析、功能基因组学
2
背景
自高通量检测技术引入分子生物学以来,发展了复杂基因相互作用分析方法(与差异表达基因检测方法并行)。多年来,人们提出了三代基因集分析方法。
第一组被称为过度表征分析(ORA),建立于1999年[1]。各种各样的工具都属于这一类,包括简单的工具,如GOstat[2]和David[3],或者更复杂的工具,如RuleGO[4]。ORA中基因集显著性的统计评价主要是基于超几何检验、χ2检验或FISHER精确检验,从而使第一代方法简单易行。然而,ORA有两个严重的缺陷:特征分化强度的信息由于二值化而丢失(基因集中的特征只表示为差异表达的基因或非差异表达的基因);在超几何检验中,大多数情况下不能满足基因表达独立性的假设。
为了克服这些问题,第二代基因集分析方法于2003年被提出[5],被称为功能类排序(FCS)。这些方法使用所有被分析基因的信息,并根据某种度量对它们进行排序。将来自基因级别的信息进一步转化到通路级别(这一过程是每个算法所特有的),并建立每个基因集的统计意义。然而,基因集是独立分析的(如在ORA中),根据生物学知识进行基因调控的方向并不包含在内。文献中提出了几种FCS方法,如Camera[7]、PLAGE[8]和GSEA[5,9]。
同时,2004年开发了第三代方法(基于通路拓扑(PT)的方法)[10]。在结构上,这些方法类似于FCS,但它们使用通路拓扑来计算基因水平的统计信息。在这一组方法中,提出了NetGSEA[11]、CEPA[12]或hybrid approach EnrichmentBrowser[13]等方法。
尽管第三代方法似乎最符合分子水平生物学的复杂性,但它们也有一定的局限性。最主要的一个是,真正的通路拓扑取决于细胞周期的阶段,细胞类型或特定的条件,但这方面的信息现在很少有。此外,它们需要更大的计算资源。(高大上的统计往往实现起来比较困难)
尽管存在上述缺点,但第一代和第二代方法仍是常用的方法。其中,GSEA方法似乎是最受欢迎的,在Google Scholar[9]上引用了1万多篇原创文章[9](2016年才被引用了1000多次)。研究人员将这一程序应用于各种基因组研究,包括lncRNA[14]、micRNA[15]或复杂疾病的系统生物学[16、17]。GSEA是为基因表达数据的分析而构建的,但在处理SNP数据(如GSEASNP[18]、MAGENTA [19]和I-GSEA4GINS[20])或RNA-seq数据(如SeqGSEA[21])方面有一定的扩展。
基因集富集分析算法的第一个实现是由原始概念的作者在Java中创建的[22]。该应用程序最大的优点是用户界面友好,有几种不同的排名方法,还可以访问来自Broad研究所的基因集资源。在名成为GSEA-P-R的R包中提供了相同的功能。最近,出现了新的实现方法。在RapidGSEA软件套件[23]中,他们提出了两种基于置换的GSEA工具,使用在CUDA指定的GPU(CudaGSEA)或多核CPU(OmpGSEA)上的并行计算。他们通过计算局部偏差引入了一个简单的基因排序指标。在这两种实现方法中,都不可能使用其他基因排序指标。
GSEA程序通常由基于Java应用程序[22]使用,其中需要设置并可能影响最终结果的主要参数是用于测量表型之间基因表达差异程度的排序指标的选择。在基于Java的标准应用程序中,实现了基本的度量标准,如:信噪比(S2N)、两类平均表达式的比率(比率)、T检验统计量(T检验)或用于定量研究的Pearson相关系数。然而,研究人员还使用了其他基因排序指标,如来自t检验的对数转化p值[24]、对应于Welch t检验的单边p值的高斯z值[25]、微阵列的显著性分析[26]。新的排序指标与预先排序的GSEA程序相结合,进行基因排列以获得富集分数分布。然而,这种类型的排列是不推荐的,因为它失去了基因-基因的相关性,所以表型取样更合适[9,27]。
这里,我们对GSEA的多个排序度量进行了复杂的比较,包括在标准Java应用中实现的排序度量和在高通量数据特征选择中成功应用的新度量。为了评估测试排序指标的有效性,我们提出了两个独特的、统计上合理的度量,这两个度量是通过对[28]引文中提出的度量进行修改而创建的。所提出的测度是伴随着计算时间的,可用于任何其他比较研究。到目前为止,还没有对基因集富集分析中的排序指标进行检验的研究[29-31],但是我们在这里使用的是具有强大表型排列、大量数据集和基因集分析统计质量度量的各种排名。最后,我们在matlab中实现了gsea方法,并将其命名为MrGSEA (matlab指标gsea-https://github.com/ZAEDPolSl/MrGSEA).)。该实现包括所有经过测试的排序指标,并保留了使用最强大的表型排列实现新的排序指标的可能性。此外,该实现具有并行计算能力。所有这些都可以改进和扩展现有的基于Java的解决方案[22]或基于CUDA的解决方案[23]。
3
方法
数据集
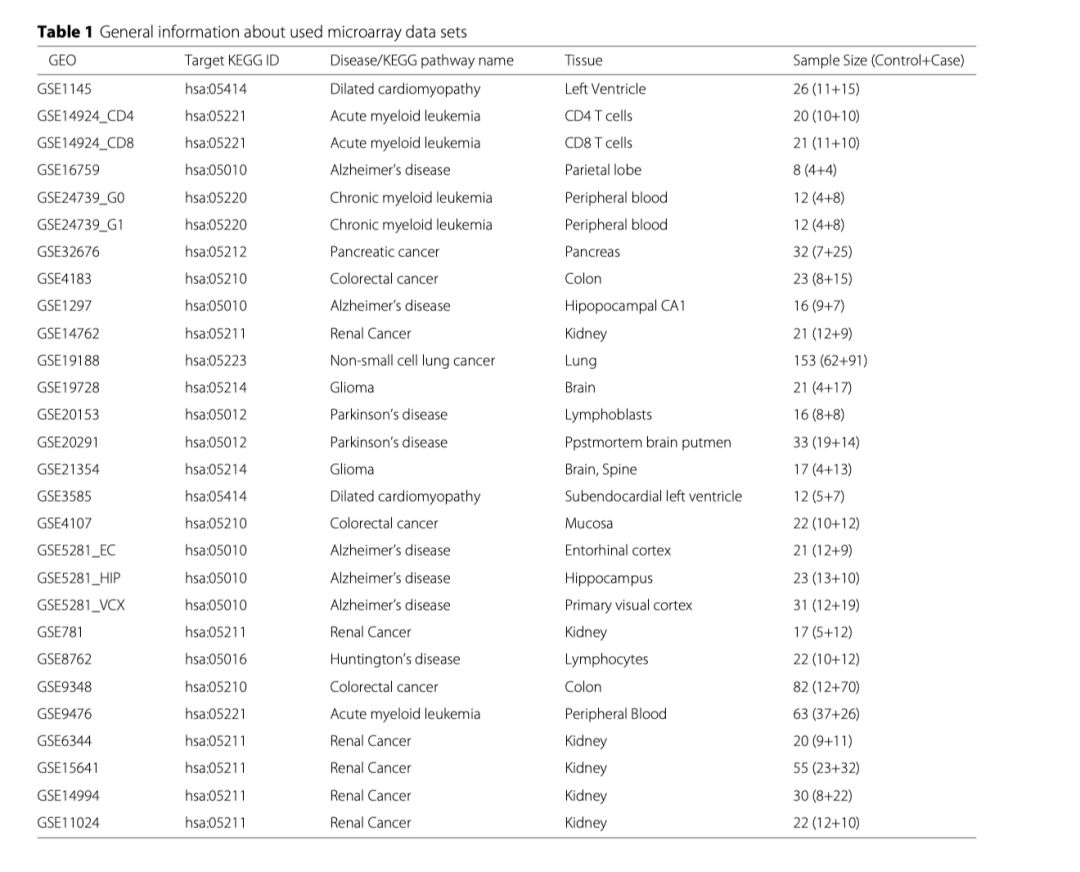
使用两个BioConductor软件包的数据集,公开可获得的微阵列数据集。在这两个数据集的集和中,对每一种疾病都指定了来自“京都基因和基因组百科全书”(KEGG)[32]的目标通路,该通路提供了关于所测试的基因排序指标的效率的主要信息。根据KEGG疾病资源,对疾病的目标途径进行了定位,指出了每种疾病的主要途径。对于每一种疾病,选择与疾病同名的通路作为靶通路,例如,当研究对象为肾癌时,靶通路为hsa:05211,称为肾细胞癌。第一个数据收集由KEGGdzPathwaysGEO软件包[6]提供,其中包括24个微阵列数据集,第二个数据收集由KEGGandMetacoreDzPathwaysGEO package软件包[28]提供,其中包括18个微阵列数据集。从这两个数据集的集合中,使用配对研究设计和MetaCore通路识别的数据集被删除。此外,以前在[33]中使用的肾透明细胞癌(CcRCC)的数据集从基因表达总汇[34](ID:GSE6344,GSE15641,GSE14994,GSE11024)下载并包括在分析中。对于KEGGdzPathwaysGEO软件包,通过保持探针集的p值最小,以及对于KEGGandMetacoreDzPathwaysGEO和cRCC数据集,通过在所有样本中保持最高的平均表达水平,删除了对基因的重复探针集[6]。对每组数据进行Lilliefors检验,发现非正态分布基因所占比例。所使用的28个数据集的详细说明见表1。作为一个基因集合KEGG通路通过KEGGREST软件包(11/2015更新)使用,由299条不同的通路组成。
基因集富集分析方法
由Subramanian等人提出的基因集富集分析方法。[9]由于分析的实验条件之间基因表达的差异,[9]仍然是用来测试通路(基因集)可能失调的最流行的方法之一。它被归类为第二代方法,与样本随机化相竞争(关于被测试假设的更多细节见[27])。GSEA方法的基本思想是使用加权Kolmogorov-Smirnov检验统计量来检验基因在基因集中的分布(根据已建立的排序度量)是否与均匀分布不同。为了建立感兴趣的基因集,富集分数被计算为基因g的命中与标记为Phit的基因集S之间的最大偏差(Eq。1)和基因g外的基因集S标记为Pmiss (Eq.。2):
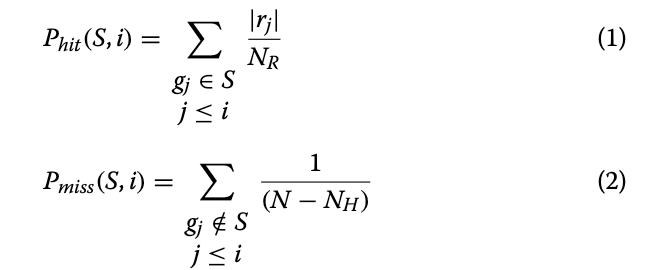
其中NR是基因集S中所有基因排序指标的绝对值之和,NH是一个基因集的大小,N是一个被分析基因的总数,r是一个排序指标的值,表示实验组之间基因表达的差异有多大。i和j表示基因等级排序列表中的位置。观察到的富集分数的意义是通过排列测试来评估的。在GSEA方法中,可以执行两种类型的排列:按样品排列或按基因标记排列。由于只有样本排列类型允许保持基因相关结构,这是推荐的[27],因此在本文的工作中只考虑了这种方法。最后,为了调整估计的富集分数以适应基因集合大小的变化,计算了归一化的富集分数。在本研究中,标准化富集评分的p值被用来作为通路富集的度量。
排名指标
我们将16个排名指标分成两组进行比较。表2列出了所有测试指标的详细公式。第一组由标准GSEA的Java应用程序中可用的指标组成[22]:信号噪声比(S2N;GSEA中的默认度量)、信噪比绝对值(|S2N|)、类间表达手段差异(Difference)、两类表达手段之比(Ratio)、比值的log2(Ratio)和T检验统计量(T检验)。第二组由来自特征选择领域的排序指标组成,用于高通量生物学实验中差异表达基因的发现。前两个指标基于适度Welch检验统计量(MWT及其绝对值|MWT|),使用t检验过程中的加权合并和未合并标准误差计算,并通过估计基因间的基因水平方差进行调整[35]。接下来两个排序指标使用非参数检验统计量:秩和(SOR)和BWS检验统计量(BWS)[36]。这两种方法在[31]之前都已在GSEA中使用过。与其他方法不同的是,这些度量没有对基因表达数据的分布做出假设。SOR是基于属于某一特定实验班的基因的排名之和。BWS检验是基于两个经验分布函数之间的差值的平方,用各自的方差加权,并用每个类的B统计量的平均值来近似。Neumäuser表明,与Wilcoxon检验[37]相比,BWS提供了更精确的I型错误控制和更大的功率。进一步的两个度量是从ReliefF法得到的,该算法对每个基因评估一个权重(从1为最好到-1为最差)。权重表示基于最近邻距离估计的类之间的最佳分离[38]。
对于每个权重,绑定的排名被指定为第二个基于ReliefF的度量(ReliefF排序)。此外,还包括加权平均差分法(WAD)及其绝对值(|WAD|)。作者认为,WAD在识别差异表达基因方面具有更好的敏感性和特异性,与标准的平均差异或倍性变化相比,WAD在识别差异表达基因和更稳定的顶级基因列表方面具有更好的敏感性和特异性[39]。第二个被测排序指标是倍数变化排名统计量(FCROS),它基于由样本组之间的两比较得到的倍数矩阵计算出的截断均值[40]。最后,我们使用最小显著性差(MSD)[41],它被定义为从无变化(零)开始的倍数变化对数(LogFC)估计的置信区间(CI)。

这可以解释为对logFC最悲观的估计,它仍然在95%的CI之内。MSD度量的值表明,在95%的情况下,logFC将至少具有此大小。MSD的负值表示logFC为零在CI内。虽然我们的MSD的实现是参数化的,但一般来说,CI的计算可以在非参数的框架下实现。
运行
GSEA方法是在64位MATLAB R2016a编程环境下实现的。测试的所有排名指标均可用。利用MATLAB的内在功能,使用外部排序度量方法是可行的。对于每个排序度量,软件通过使用排列测试来计算对应于不同表型之间表达差异的p值。富集分数分布可以通过基因标签或表型的排列来估计。此外,还有一个功能可以从包含成基因集的基因的Excel文件中创建自定义基因集数据库。富集分析的结果以MATLAB变量、Excel文件和PNG图像的形式存储。
该算法采用复制工作者的想法进行并行化。由于GSEA方法的性质,每个线程分别计算不同基因集的富集分数及其分布。由于每个基因集中的基因数目不同,导致计算时间不同,因此该解决方案提供了最快的结果。并行执行的任务数量取决于可用的处理器内核数量和MATLAB软件许可(使用并行计算工具箱最大12个线程)。带有示例数据集和演示脚本的源代码可以从https://github.com/ZAEDPolSl/MrGsea[42]免费下载。
4
实验设计
计算实验是在收集28个微阵列数据集的基础上进行的,每个数据集都有一个目标通路。对16个GSEA排序指标进行了两个评分:1)替代敏感性-目标通路标准化富集评分统计数据的p值(越小,越好)(参见[28]s1注);2)假阳性率(FPR)-在5%的显著性水平上发现的假阳性率(越接近5%,越好)。为了评估FPR,对每个数据集的原始表型进行置换,形成50个独立的数据集合。为了估计给定的排序度量的总体灵敏度,使用了多重检验的Storey’s方法中的保守估计πˆ0[43]。πˆ 0表示真正空测试的比例,期望所有p-值将遵循均匀分布,其中1-πˆ0是真正替代测试的比例。为了估计排序指标的总体FPR,取平均FPR(观测水平)与5%(预期水平)的绝对偏差。此外,还对每个排序指标的计算负荷进行了评估。获得的分数(总体灵敏度、总体FPR、计算负荷)被归一化到一个尺度上,根据基因集富集的总体性能,使用欧氏距离的k-means程序来划分排序指标。
k-means算法中的聚类数是使用Dunn指数[44]来确定的。最后,为了获得最好的排序指标,在以下情况下测试了其对样本大小的稳健性:将最大数据集(GSE19188)以分层的方式随机分为不同的样本大小集合(10、20、30、40、60、80和100),每组样本大小分别为10次、20次、30次、40次、60次、80次和100次。对于每一种情况,计算了特定方法的基因集富集的替代灵敏度和FPR。
5
结果与讨论
基因集分析的总体敏感性和FPR
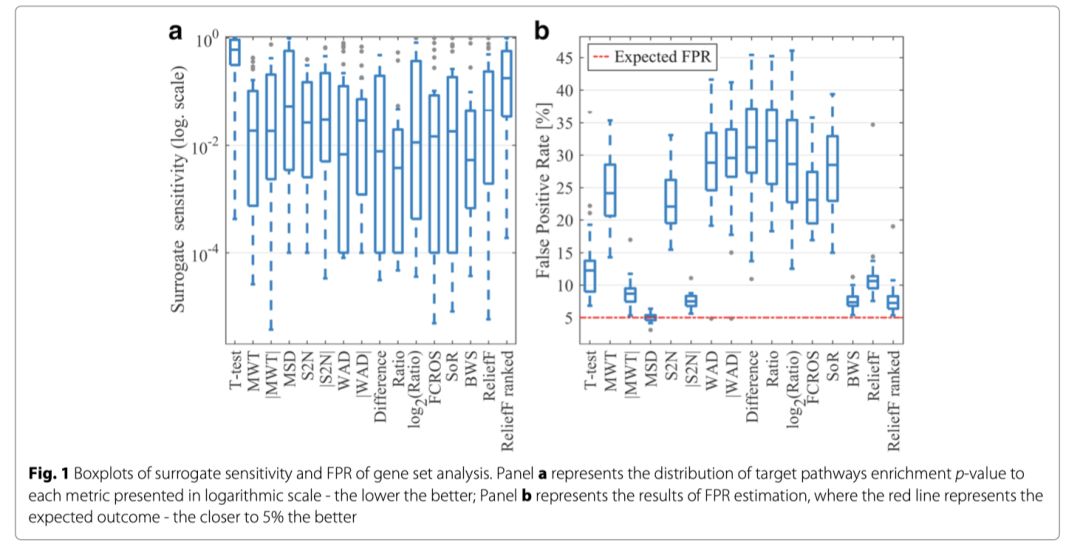
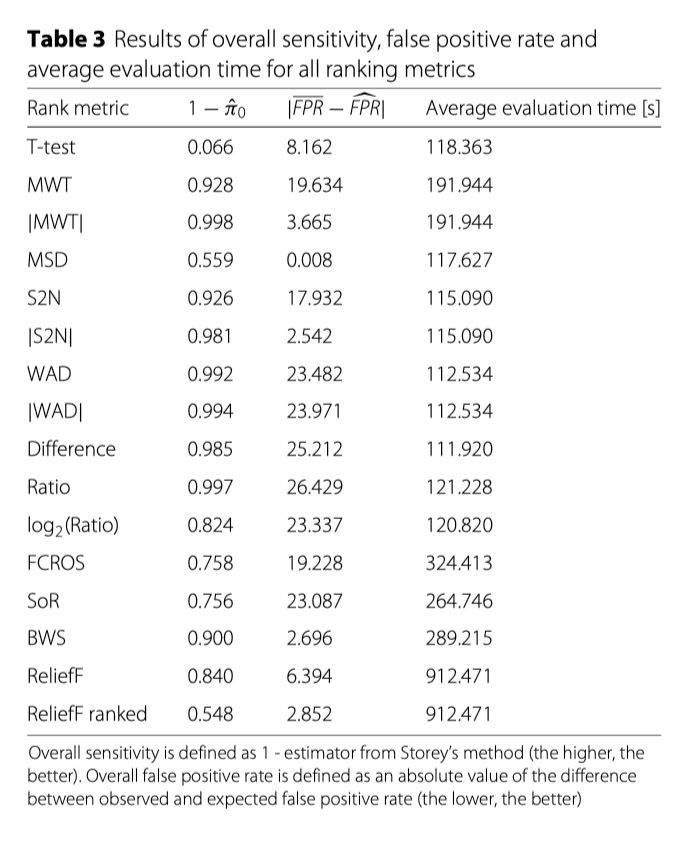
对28个数据集中的每一个都进行了GSEA方法,其中包含1000个表型排列和16个不同的排序指标。图1A中显示了代表替代灵敏度的目标通路的p值。图1B中显示了代表FPR的显著富集基因集的平均百分比。在所有的数字中,排序指标是从最参数化的统计数据,通过非参数的数据挖掘方法。如图所示。1A,我们可以区分两个排序指标,即Ratio和BWS,它们具有最低的替代灵敏度中值和相对较小的分布范围。最差的结果是观察到的T-检验统计量和ReliefF排序指标。对于FPR,观察到具有较低值的七个指标组:T-test、|MWT|、MSD、|S2N|、BWS、ReliefF 和 ReliefF 排序。为了找出排名指标的总体表现,对两个分数的预期结果进行了估计:总体灵敏度的1-FPR的0估计量[43],以及平均πˆ和预期FPR之间的差值的绝对值(表3)。理想情况下,对于引入的定义,排序指标应具有较高的总体灵敏度和较低的总体FPR值。此方法突出了四个排名指标:|MWT|、|S2N|、BWS和ReliefF。以较差的FPR估计为代价,比率度量具有很好的总体灵敏度,而ReliefF法分级度量和MSD则相反。此外,每个数据集上的每个排序指标的替代灵敏度和FPR的详细结果在附加文件1中显示。
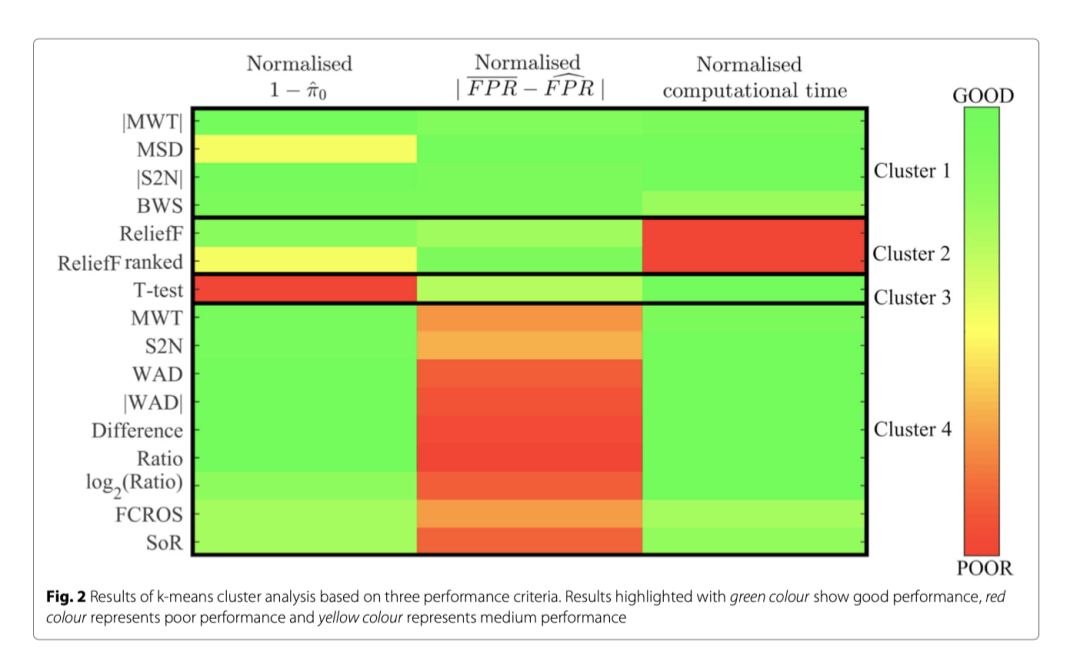
建立最佳排名指标
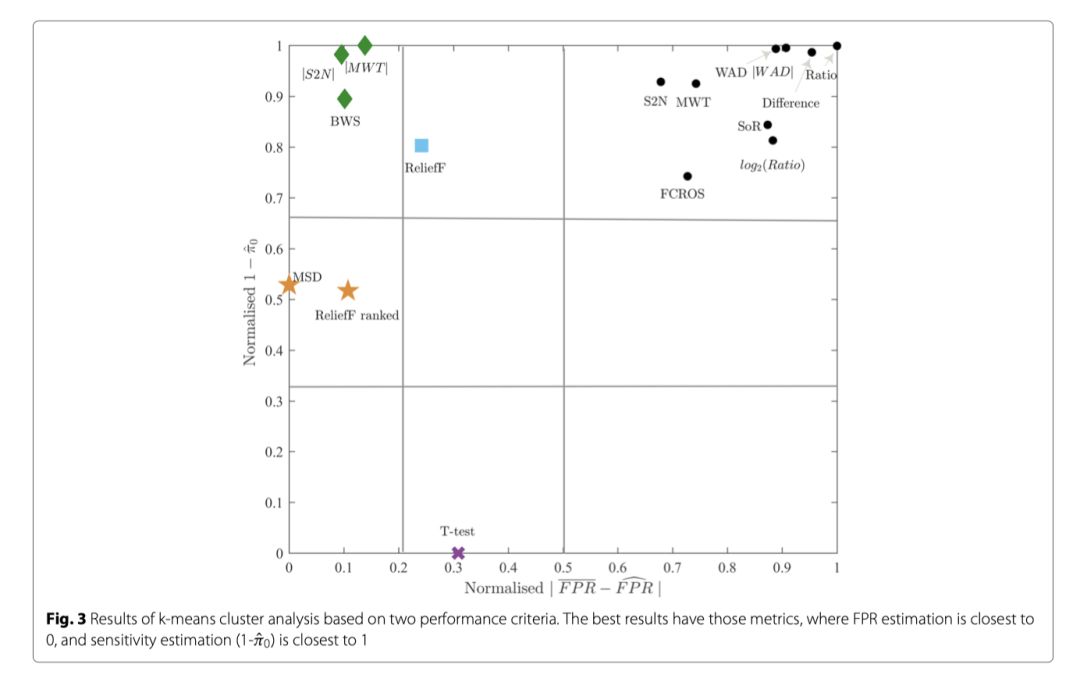
对于所有的排序指标,计算负荷是计算给定方法实际应用的一个重要指标(对每个指标和数据集的详细评估包含在附加文件1中)。为了寻找具有最佳总体灵敏度、总体FPR估计和较低计算成本的指标,采用了欧氏距离的k-均值聚类方法。每个分数的估计量被归一化到范围[0,1],以避免偏袒单一分数。Dunn指数表明四个簇是聚类的最优解。图2中给出了对4个簇使用k-means的结果。最相关的集群是那些计算负载和总体FPR较低、总体灵敏度较高的集群。聚类1满足这三个条件(图2)指标:|MWT|、MSD、|S2N|和BWS- 绿色优势。在这四个指标中,|MWT|具有最高的总体灵敏度,而MSD在测试数据收集方面表现出最低的总体FPR和较低的计算负载。在此组中,只有|S2N|在最初的基于GSEA Java的实现[22]中可用,这是由于[45]中获得的结果而添加的。对于图中所示的其他簇。我们可以观察到至少一种被测指标(红色)的劣值。此外,k-means过程仅应用于总体灵敏度和总体FPR估计量,以显示具有最佳排序指标的另一组,当计算负载不重要时(图3)。正如可以观察到的,|MWT|、|S2N|和BWS位于一个结果最好的集群中,但这一次K-means程序也区分了另外两个得分较低的有趣集群。第一类包括具有中等FPR估计和较好的总体灵敏度的ReliefF法度量;第二类包括MSD和ReliefF法排序的度量,其总体灵敏度较低,但FPR估计较好。
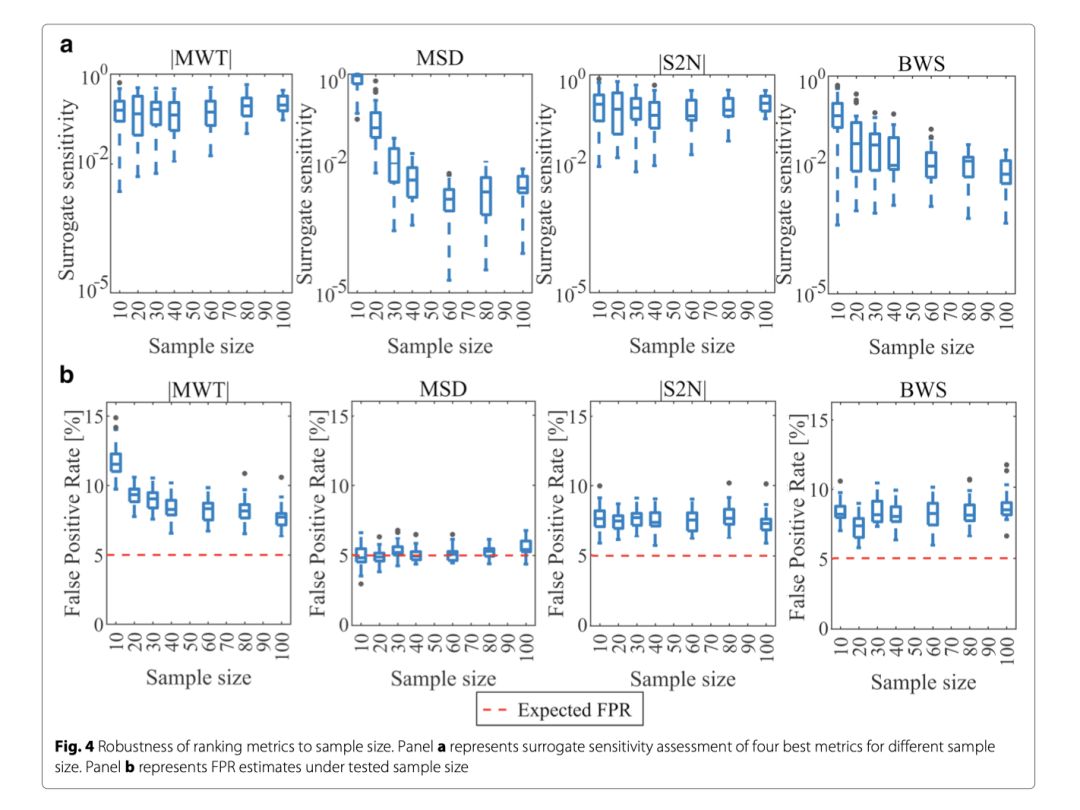
对样本大小的鲁棒性
对四个最佳排序指标(|MWT|、MSD、|S2N|和BWS)进行了测试,以确定通路富集分析相对于分析数据集中样本数量的稳健性。对每个指标和不同样本大小的替代敏感度和FPR进行了评估(图1)。4).。|MWT|和|S2N|这两个指标具有类似的替代灵敏度水平,与样本大小无关。对于较大的样本大小,BWS和MSD指标显示出更好的结果。对于BWS,它与B统计量的经验估计方法有关。在MSD的情况下,计算logFC CI的标准误差估计较弱。在FPR的情况下,可以观察到它对于所有度量和样本大小都是恒定的。|MWT|方法是唯一的例外,在极低的样本大小下,得到的FPR较高。对排序指标对样本大小的稳健性分析表明,|NWT|和|S2N|给出了稳定的结果,但对于较大的实验,得到的替代灵敏度比BWS和MSD要差。
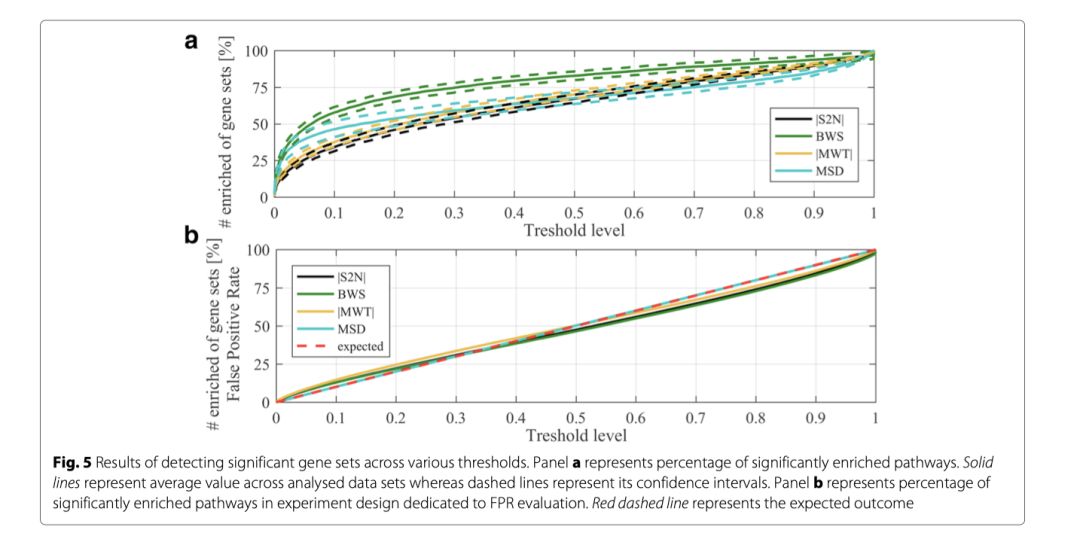
通路富集的精确度
最后,确定了不同显著性水平的富集途径的百分比。图5a显示了测试数据收集的平均已发现通路的百分比,其置信区间为95%,而图5显示的是已发现的测试数据收集通路的平均百分比。图5b表示样本标签随机排列后富集通路的平均水平(假阳性估计)。可以看出,BWS检验排序标准在不同的阈值水平上给出了统计显著性基因集的最高比率,将假阳性结果保持在预期水平。|S2N|、|MWT|和MSD的结果非常相似,彼此之间在0.05显著水平上没有差异。对于这三种方法,假阳性的预期数量也是保持不变的。这些结果表明,BWS排序指标不仅可以发现p值较低的预期目标通路,而且与其他指标相比,可以检测到更多的不规范的通路,并且错误的结果被接受的数量更多。在28个数据集中,还需要进一步的研究来确认BWS发现的额外的重要基因集与研究中的表型之间的联系。
最佳排名指标的选定属性
四个最佳排序指标中的三个(|S2N|、MWT和MSD在当前执行中)是使用算术平均值和标准差计算的。由于这些都是参数统计,所以提出了两个假设:数据分布的正态分布和不存在离群值。在利用高通量生物学技术获得基因表达的情况下,通常不满足正态分布的假设,并且经常出现离群值。因此,这可能导致通路富集分析的功耗降低。这项研究中使用的数据收集的特点是基因表达的分布并不严重倾斜(见附加文件2),因此参数方法所描述的缺点并不明显。在0.05的显著性水平上,平均约有25%的基因具有非正态分布,但当我们将显著性水平设置为0.01时,这个数字下降到11%。值得注意的是,MSD是否取决于参数假设,取决于具体的实现。另一方面,BWS检验统计量没有关于数据分布的假设,因此允许对每一种生物数据使用该度量。在一组被分析的数据集中,非正态分布基因的比例与使用BWS获得的总体灵敏度增益相比,|MWT|和|S2N|显著相关(p值分别为1.16E-4和1.03E-4)。BWS与MSD相比,总体灵敏度无明显提高(p值=4.56E-1)。这些结果与Neutäuser和Senske[37]得出的结果类似。它们还表明,在对称分布的情况下,BWS具有与参数检验相似的能力。
GSEA方法允许检测基因集合中的失调方向(上调或下调)。结果取决于所使用的排序指标的类型。当高正、低负值的排序值代表差异表达的基因时,我们可以检测到大部分基因表达上调或下调的基因集。当缺少由于表型引起的基因表达方向变化的信息时,GSEA只允许检测不受调控的基因集(尽管表达方向不同)。第二种情况是所有四个最佳排序指标。这些发现与生物学知识是一致的,在相同的途径中,可以观察到基因的上调和下调。然而,在一些生物学实验中,只用上调或下调的基因来富集通路是可取的,因此在使用GSEA时,研究人员必须了解它的性质。
这里可以回过头看看:GSEA分析合理性讨论
|S2N|和BWS指标的一个共同缺点是,每个基因的p值(表现出表型之间的统计差异)只能通过排列检验[46]来估计。众所周知,当精确的p值很低时,排列测试估计是非常耗时的。通过使用|MWT|和MSD排序指标,可以在不增加任何计算成本的情况下获得精确的p值。
最后,MSD排序指标需要更多的排列(在我们的分析中超过10,000个),才能正确地估计基因集富集的p值。这一现象是由于MSD度量值存在负值而引起的,表明log倍数变化不大。 当我们进行标记置换时,表型内基因表达的方差增加,导致MSD值为负的基因数量(以及更多的负归一化富集评分的通路)的增加。在GSEA中,为了计算给定通路的p值,只使用归一化富集分数分布的正或负部分,对应于观察到的归一化富集分数的符号。在所有情况下,在使用MSD时,置换数据的归一化富集分数的分布包含的正值比负值少得多,因此为正NES建立适当的p值需要更多的排列。然而,这一弱点也以降低假阳性率的形式出现积极的后果。此外,MSD的定义允许即使在复杂的线性模型或对比的情况下也能实现,从而提供了更大的灵活性。
结论
在使用28个微阵列数据集的情况下,对GSEA算法中16个排序指标的基因集结果进行了对比观察。在计算负荷的同时,采用灵敏度和FPR的统计方法对各指标进行检验。在所有测试方法中,有4种结果较好,即|MWT|、MSD、|S2N|和BWS。在测试指标中,|MWT|的总体灵敏度最好,而MSD获得了最佳的总体FPR估计。在四个最佳指标组中,|MWT|、MSD(在当前实施中)和|S2N|基于参数估计,在不满足此假设时应谨慎使用。与其他指标相比,BWS显示了更大的样本容量和非正常的基因表达分布的更好的结果。它还检测到更丰富的通路,将错误的发现保持在合理的水平,这可能会提示新的发现。我们表明,在GSEA的情况下,选择排名度量是很重要的,它的作用是不可忽视的。适当设置排序度量可以改进[28]中批评的GSEA方法的FPR估计。尽管如此,使用任何排名指标都是可能的,但研究人员必须意识到这项研究中可能存在的弱点。在所附的MATLAB实现GSEA计算被有效地并行化,从而有机会轻松地修改脚本以满足研究者的期望。与现有的实现相比,MrGSEA提供了更高的灵活性和功能,表现为大规模的排序度量(包括自己的)、使用定制的基因集数据库和两种不同类型的排列。
本文翻译自文献:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1674-0