从神经系统变性病看疾病诊断的演变和认识重构(3)--基因诊断及其临床价值
编者按:我们所处的医学时代,是一个各种认识、定义和观念在不断重组的时代,从传统意义上的临床诊断过渡到最终的精准诊断是一个让人迷惑和焦虑的过程,如何厘清疾病诊断在不同维度的表现和关联,不但要及时把握最新的研究进展,还需要自己用心归纳和总结,以免在各种诊断“林立”的现在失去对疾病的决策和判断!从本期开始,本公众号有幸邀请青岛市立医院神经科郁金泰教授以神经系统变性病为例,来探讨疾病诊断的演变和认识重构(重组)!

疾病的基因诊断和其临床价值
FTLD的基因诊断
FTLD患者有很强的家族聚集性特点,40-50%的患者有阳性家族史,其中10-50%呈孟德尔式常染色体显性遗传。其中,bvFTD患者最常出现阳性家族(45%),尤其是伴有ALS时(60%),而svFTD的遗传性最低(<20%)。分子遗传学研究发现 了5个可以引起FTLD的致病基因(图11): 微管相关蛋白-tau(MAPT)基因、颗粒蛋白前体(GRN)基因、含缬酪肽蛋白(VCP)基因、带电荷的多囊泡体蛋白2B(CHMP2B)基因和最近确定的C90RF72六核苷酸重复扩增。

图11. FTLD的主要致病基因。(参考文献6)
现有研究发现,MAPT、GRN和C90RF72为家族性FTLD最常见的致病基因,它们的突变可以至少解释17%的家族性FTLD患者。而VCP和CHMP2B的突变相对比较罕见,尽可解释不到1%的家族性FTLD患者。此外ALS的常见致病基因TARDNA结合蛋白43(TARDBP)基因和肉瘤融合蛋白(FUS)基因突变,偶尔也可以呈现FTLD临床表现,但非常罕见。
1996年在美国举行的一次国际会议上,总结报道了13个FTLD家族,这些家族患者的主要表现为早老性痴呆,临床上表现为行为、认知和帕金森综合征相关运动症状,连锁分析定位于17q21-22,并命名为额颞叶痴呆合并帕金森综合症(FrontotemporalDementia and Parkinsonism linked to chromosome 17 ,FTDP-17)。1998年发现位于该区域的tau蛋白编码基因MAPT是这些家族最主要的致病基因。到目前为止已发现了44个不同的MAPT致病突变位点。
虽然在散发性FTLD患者中MAPT突变很罕见,在家族性FTLD患者中约5-20%可以发现MAPT突变,并且突变主要位于9、10、11和12号外显子上。随着二代测序的发展,2006年发现GRN基因的功能缺失性突变也是FTDP-17的常见病因。GRN突变大约和5-20%有阳性家族史的患者有关,和1-5%散发性FTLD有关(参考文献6)。
在17q21相关FTLD发现其致病基因为MAPT和GRN后,FTLD的遗传学研究领域转向了染色体9p相关的家族性FTLD。FTLD-9主要表现为ALS或FTLD-ALS。2011年,Neuron杂志同期刊登了来自欧洲和美国两个课题组的研究发现,这两个课题组同时报道了染色体9p区域的C9orf72基因GGGGCC六核苷酸重复是FTLD-9致病原因(DeJesus-Hernandez et al. 2011,Renton et al. 2011)。
C9orf72基因GGGGCC重复扩增是欧美家族性FTLD最主要的原因,大约和25%的FTLD患者和5%的散发性FTLD患者相关。不过,国内的研究发现,C9orf72基因GGGGCC六核苷酸重复扩增在汉族等亚洲人群中非常罕见,这也体现了不同种族的遗传异质性。
此外,许多神经变性病都要家族聚集的现象,但是这种家族聚集并不一定表现为家族性遗传性疾病,同一个家族中可以出现不同表型,甚至不同病理类型的变性病,如同一个家族中,既有PD,又有PSP、AD患者等,这也进一步支持了这些变性病有许多相同的共性和病理发生机制,目前也有大量的这方面文献的报道。
和其它神经变性病的遗传学研究现况不同,FTLD目前的遗传学研究发现主要集中于外显率非常高的罕见突变(图12),它们大多和家族性FTLD相关,而对于散发性FTLD的GWAS研究目前仅报道TMEM106B和GRN基因,这可能和FTLD的临床表型和病理分型异质性太大有关。

图12.FTLD的遗传学现况和未来发展趋势。(参考文献7)
基因诊断的临床价值
基因诊断方法就是利用现代分子生物学和分子遗传学的技术方法,将样本(血液、体液、组织)从人体中取出后进行基因检测而进行诊断,从而对疾病作出诊断的方法,属于病因诊断。随着遗传学的发展,尤其相关基因检测技术的进步,基因诊断近些年的风头有超越病理诊断的趋势,尤其精准医学理念的提出进一步炒热了基因诊断。目前对于家族性遗传性疾病,基因诊断的地位已经超越了病理诊断的地位,处于疾病诊断的金字塔顶端。而对于散发型多基因复杂性疾病,近些年临床医师、遗传专家和计算机专家也在积极的合作,试图建立一套基因多基因评分的预测和诊断模型。
随着二代测序快速发展尤其测序变得越来越便宜,基因测序在临床上的应用越来约广泛,然而很多医生面对测序发现的许多相关突变不知道如何解读,常常搞不清到底哪个是致病突变,哪个只是疾病不相关的突变携带。对于外显率100%的家族性疾病,目前基因诊断已经可以代替病理诊断,作为确诊金标准,如HD、肝豆等。但是对于散发病例来说,基因诊断要慎之又慎。
既往有文献报道不足以证明突变的致病性,需要结合受检者具体临床表现并结合相关研究信息进一步分析尤其对于散发病例,不要对于散发病例,相关基因测序后发现个有文献报道的致病突变,并认为该突变为该患者的de novo致病突变。de novo的致病突变需要全面的证实,如父母是亲生父母,父母不存在该突变,家族中其它成员也不存在该突变等等。这也是为什么我们明明临床症状和符合某中疾病的诊断,基因检测也发现了以往文献报道过的突变,为什么投稿时审稿人还是不相信和接受我们的报道,让我们补充很多其它相关的内容。
尤其对于散发性病例,当临床诊断和基因诊断不一致时,肯定还需要病理诊断来确诊。在变性病中,当临床诊断和病理诊断不一致时,我们拿病理诊断作为金标准,将该临床诊断作为该疾病的atypical或clinical variant,但是对于基因有时候就不能这样来定义了。所以说对于一个散发病例来说,笔者认为其诊断的确定性,从塔尖到塔底应该是:临床-病理-基因完全一致>病理-基因一致>临床-病理一致>病理诊断>临床+基因一致>临床诊断>基因诊断。上述的排序,应该也和期刊接受临床个案或临床病例投稿的接受或认可程度相一致,越位于塔尖的病例分析越容易发表。
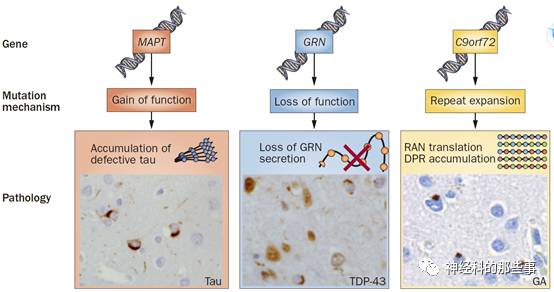
基因诊断的优势是可以在生前就明确相关疾病的病因和潜在的病理,其不仅对优生优育、疾病的遗传咨询和预防有重大价值,还对药物临床试验的设计、疾病的进程和临床转归预防等方面均有重大临床价值。此外,疾病相关致病或风险基因的发现,对疾病发病机制的阐明(图13),潜在细胞分子机制(图14)和相关疾病调修药物的研发(图15)等都有重要的科学意义。
图13.三种FTLD的基因、突变机制和病理之间的关系。(参考文献8)

图14. FTLD发病相关基因编码蛋白的细胞分布和功能通路。(参考文献7)

图15.靶向APOE研发AD调修药物。(参考文献9)
大家目前做神经变性病基础研究的相关转基因动物模型,如APP/PS1转基因小鼠、P301转基因小鼠,也都是从相关家族性疾病的致病基因发现研发出来的。可以想象,将来基因诊断会变得越来越热,尤其在成功的研发出来相关的疾病调修药物后,或者基因治疗发生革命性的的进展。
参考文献
6. Tang SS, Li J, Tan L, Yu JT. Genetics of FrontotemporalLobar Degeneration: From the Bench to the Clinic. J Alzheimers Dis. 2016 Apr19;52(4):1157-76.
7. Pottier C, Ravenscroft TA, Sanchez-Contreras M, RademakersR. Genetics of FTLD: overview and what else we can expect from genetic studies.J Neurochem. 2016 Aug;138 Suppl 1:32-53.
8. van der Zee J, Van Broeckhoven C. Dementiain 2013: frontotemporallobardegeneration-building on breakthroughs. Nat Rev Neurol. 2014 Feb;10(2):70-2.
9. Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer'sdisease: an update. Annu Rev Neurosci. 2014;37:79-100.