地中海贫血基因检测,你知道多少?
地中海贫血(thalassemia)也称海洋性贫血,珠蛋白生成障碍性贫血,是由于遗传的基因缺陷导致血红蛋白中至少一种珠蛋白合成缺乏或不足引起的贫血或病理状态。因为其基因缺陷类型复杂多样,所以珠蛋白缺乏的类型、数量也各有差异,导致的临床症状也表现不一。
地中海贫血是一组遗传性疾病,根据缺乏的珠蛋白链的种类命名,α珠蛋白链缺乏者称为α珠蛋白生成障碍性贫血,即α地中海贫血;β珠蛋白链缺乏者称为β珠蛋白生成障碍性贫血,即β地中海贫血。根据珠蛋白链缺乏的程度分为完全无生成的α/β地中海贫血和部分生成的α/β地中海贫血。
一、α地中海贫血
健康人α珠蛋白链的合成是由第16对染色体上两对联锁的α珠蛋白基因所控制,这四个α珠蛋白基因继承自父母双方各两个。若继承的α珠蛋白基因缺乏或缺陷,则导致α珠蛋白链的合成速度明显降低或几乎不能合成。根据程度不同可分为四种。
1,一个α基因异常:患者无血液学异常表现,称为α珠蛋白生成障碍性贫血静止型。平常无症状,血象无特殊表现,仅在出生脐血或出生八个月内血液中Hb Barts(γ链形成四聚体γ4)轻度增加(小于2%)。
2,两个α基因异常:红细胞呈小细胞低色素性改变,称为α珠蛋白生成障碍性贫血标准型。无症状或轻度贫血,出生时Hb Barts可占5%~15%,但几个月后消失。
3,三个α基因异常:双重杂合子,有代偿性溶血性贫血表现,多余的β链聚合成为HbH(β链形成四聚体β4),即HbH病。患者血象可出现小细胞低色素改变,靶形红细胞增多,血红蛋白电泳出现HbH和Barts带,大部分细胞中可见HbH包涵体。
4,四个α基因异常:完全无α珠蛋白生成,为纯合子。临床表现为胎儿水肿综合征,胎儿期无HbF,多余的γ链聚合成Hb Barts,又称HbBarts病,胎儿多死于宫内或产后数小时。血红蛋白电泳Hb Barts大于90%,有少量HbH,无HbA、HbA2和HbF。
二、β地中海贫血
β地中海贫血的发病率高于α地中海贫血。患者从父母处遗传一个或两个异常β基因,使β链合成减少或不能合成,从而使多余的α链聚合形成不稳定的四聚体,同时δ、γ链代偿性增多,HbA2和HbF含量增加。不稳定的血红蛋白使红细胞僵硬易破坏导致无效造血。根据基因突变情况和临床症状分为三种。
1,轻型杂合子β地中海贫血:多数杂合子患者没有任何症状和贫血,血涂片可发现少量靶形红细胞,红细胞脆性试验有轻度减低,HbA2轻度增高(大于3.5%)是其特点。
2,重型纯合子β地中海贫血:父母双方均为β地中海贫血,出生后贫血进行性加重,临床表现有发热、腹泻、黄疸、肝脾肿大等。骨髓造血代偿性增生出现地中海贫血面容。相关实验室检查明显异常。
3,中间型β地中海贫血:介于两者之间,为纯合子型或双重杂合子型。
地中海贫血的血象检验可见轻重不等的贫血,红细胞大小不均,靶形红细胞和异性红细胞增多。红细胞脆性减低。骨髓象可见红细胞增生极度活跃,粒红比例倒置,呈无效性增生和原位溶血。
血红蛋白电泳对于珠蛋肽链结构异常或合成量异常的诊断有重要意义,选择适当的血红蛋白电泳可检测出各类异常血红蛋白和各种血红蛋白成分的相对含量。
随着检测技术的飞速发展,地中海贫血基因诊断技术已十分成熟完善。其所需样本量小,并可在基因层面上直接进行诊断分析,对于无症状或轻度改变的患者更具有意义,极大地提高了临床确诊度和优生优育的发展。
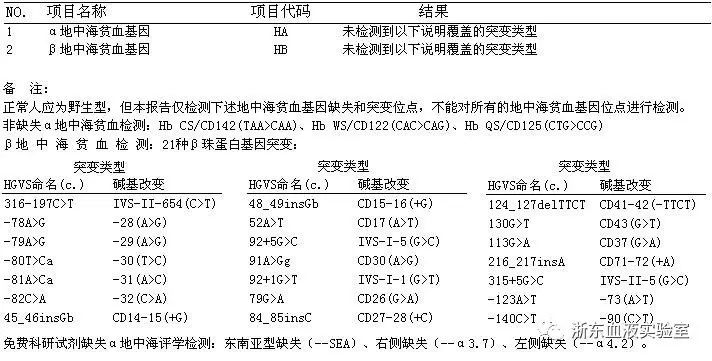
我们科室开展地中海贫血基因检测项目三年,标本量每年持续增加,今年到目前为止的标本量达到近一千两百例。我们检测α地中海基因缺陷6种,β地中海贫血21钟,涵盖绝大部分地中海基因缺陷,但由于地中海基因缺陷复杂多变,不排除少数患者是上述27种以外的小概率缺陷类型,所以我们的报告结果为具体的缺陷类型或“未检测到说明中覆盖的突变类型”,而不是“无基因缺陷”或“正常”。若检测报告为“未检测到说明中覆盖的突变类型”的患者有持续贫血症状而排除其它贫血病因,强烈怀疑地中海贫血时,可通过测序来确诊。