婴儿癫痫伴游走性局灶性发作的临床及基因突变特点
作者:尚可为、张月华、杨小玲、刘爱杰、杨志仙、刘晓燕、姜玉武、吴希如
选自:中华儿科杂志, 2016,54(10): 735-739
婴儿癫痫伴游走性局灶性发作(epilepsy of infancy with migrating focal seizures,EIMFS)属于癫痫性脑病,本病最早由意大利学者Coppola等[1]于1995年报道,称为婴儿恶性游走性部分性发作(malignant migrating partial seizures of infancy, MMPSI),2001年国际抗癫痫联盟(ILAE)将其命名为婴儿游走性部分性发作(MPSI),2010年ILAE将本病重新命名为EIMFS[2]。目前已发现7个致病基因(KCNT1[3]、SCN1A[4]、SCN2A[5]、SCN8A[6]、PLCB1[7]、SLC25A22[8]、TBC1D24[9]基因)可导致本病。该病较罕见,由于二代测序基因检测方法的应用,国际上有关本病的报道逐渐增多。但国内尚罕见本病的系统研究报道。现对9例EIMFS患儿的临床和脑电图特点以及基因诊断结果进行分析。
对象和方法
一、 对象
前瞻性收集2005年5月至2016年1月在北京大学第一医院儿科神经专业门诊及病房就诊的所有EIMFS患儿。本研究获得北京大学第一医院伦理委员会批准,批准号:2012[453],所有患儿家长均签署知情同意书。入组标准:参照Coppola[10]描述的EIMFS的临床特点进行诊断:(1)生后6个月内起病;(2)几乎持续的游走性的多种类型的局灶性发作;(3)发作期脑电图表现为多灶性放电,在一侧半球内或双侧半球之间游走,累及多个部位,临床发作与脑电图放电在时间和部位上密切相关;(4)逐渐进展的智力运动发育倒退;(5)对抗癫痫药治疗反应不佳,预后不良。排除标准:除外遗传代谢病和已知脑损伤病因导致的癫痫。
二、 方法
对患儿的临床资料进行分析,并收集患儿及其父母外周血基因组DNA。采用二代测序癫痫基因检测包(共有385个癫痫相关致病基因,包含EIMFS的候选基因KCNT1、SCN1A、SCN2A、SCN8A、PLCB1、SLC25A22和TBC1D24基因)筛查患儿的基因突变。采用一代测序的方法验证患儿及其父母相关突变位点。
患儿均有2次以上脑电图检查结果,至少进行1次3~4 h视频脑电图(VEEG)监测。9例均做头颅磁共振成像(MRI)检查及血、尿代谢筛查。
所有患儿均通过在我院儿科神经门诊、再入院或电话随访的方法获得随访资料。
结果
一、 临床特点
1. 基本情况:
共入组EIMFS患儿9例,其中男3例、女6例,围产期均无明显异常。2例有癫痫家族史,2例患儿同胞姐姐均与患儿表型相似,均因癫痫持续状态死亡。
2. 起病情况:
起病年龄为2 d至3月龄,中位年龄35 d。就诊年龄为3 d至5个月,中位年龄60 d。
3.发作表现:
均为局灶性发作,有游走性特点。发作表现为单个或一侧肢体抖动9例、双眼凝视8例、眼睑眨动6例、口唇或肢体末梢发绀5例、头眼向一侧偏转4例、嘴角或口唇抽搐4例、单个或一侧肢体僵硬3例、双手握拳3例、喉中发声3例、咀嚼、咂嘴或吞咽动作3例、流涎3例、呼吸急促2例、面色潮红1例。4例病程中有癫痫持续状态。
4.智力、运动发育:
9例起病后均有发育落后。
二、辅助检查结果
1. 脑电图:
均有异常放电,表现为游走性、多灶性放电特点;背景活动均偏慢;8例发作间期表现为大量多灶性中、高波幅棘波、尖波、多棘波、棘慢波、多棘慢波散发或连续发放,以一侧半球或某一脑区为主,1例发作间期表现为额、中央、枕、颞区左右不固定多灶、单发、连发、簇发小棘波;9例均监测到临床发作,表现为单个或一侧肢体抖动9例、眼睑眨动3例、单个或一侧肢体僵硬2例、头眼向一侧偏斜2例、口唇发绀1例。同期脑电表现为右侧或左侧一个或多个脑区起源的低波幅、低-中波幅快波,频率渐快,波幅渐高,可波及其他脑区或游走至对侧脑区,随后频率渐慢、波幅渐低。典型表现如图1。
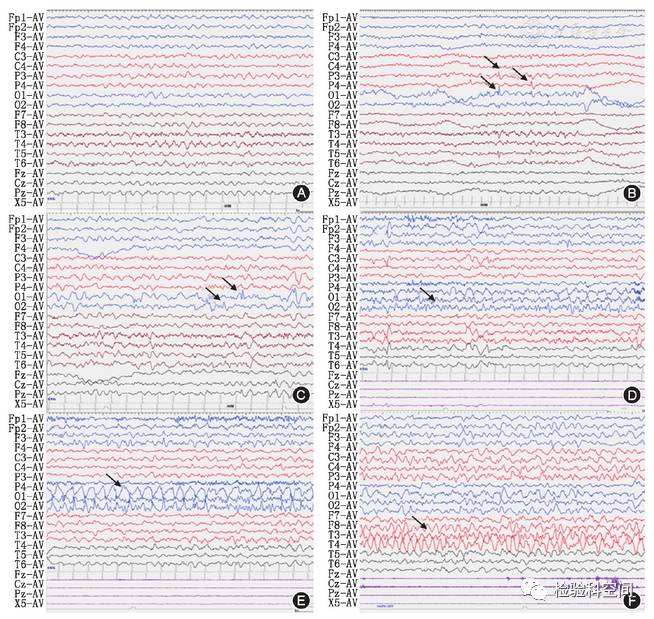
图1
婴儿癫痫伴游走性局灶性发作患儿(例1)5月龄时脑电图 A:背景节律减慢;B 、C:发作间期,显示游走性多灶性放电(箭头),后头部为主;D~F:发作期,左侧后头部起始低波幅快波节律发放(D,箭头),波幅渐高、频率渐快(E,箭头),后波及并限局于右侧颞区(F,箭头),同期双侧三角肌(X1、X2)短暂肌电暴发
2.头颅MRI:
9例患儿MRI中4例有异常。1例脑白质发育落后,大脑脑沟增宽;1例双侧额颞部脑外间隙增宽;1例双侧脑室偏大,双侧额颞区脑沟偏深;1例双侧额颞叶及小脑周围脑外间隙增宽,双侧侧脑室宽;其余5例正常。
3.血、尿代谢病筛查:
均未发现异常。
三、 EIMFS相关基因突变筛查结果
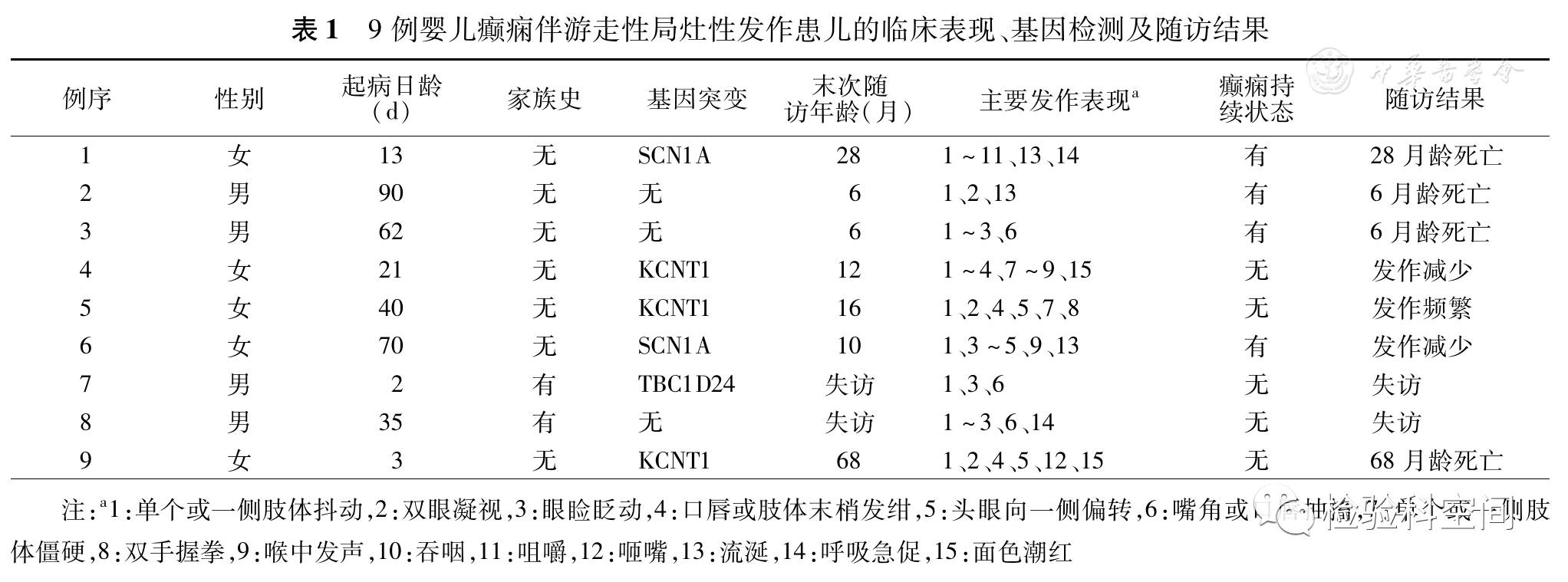
表1显示:2例患儿为SCN1A基因杂合错义突变(c.659T>A / p.Val220Asp、c.677G>A / p.Thr226Met),3例患儿为KCNT1基因杂合错义突变(c.1148G>A / p.Arg383Gln、c.1286G>A / p.Arg429His、c.1421G>A/ p.Arg474His),以上5例患儿父母外周血均未发现相同突变,均为新生突变;1例患儿TBC1D24基因突变为复合杂合突变(c.619C>T / p.Gln207*、c.866C>T / p.Ala289Va),患儿母亲携带突变c.866C>T,未获得患儿父亲外周血DNA(图2);3例未发现上述基因突变。
|
表1 9例婴儿癫痫伴游走性局灶性发作患儿的临床表现、基因检测及随访结果 |
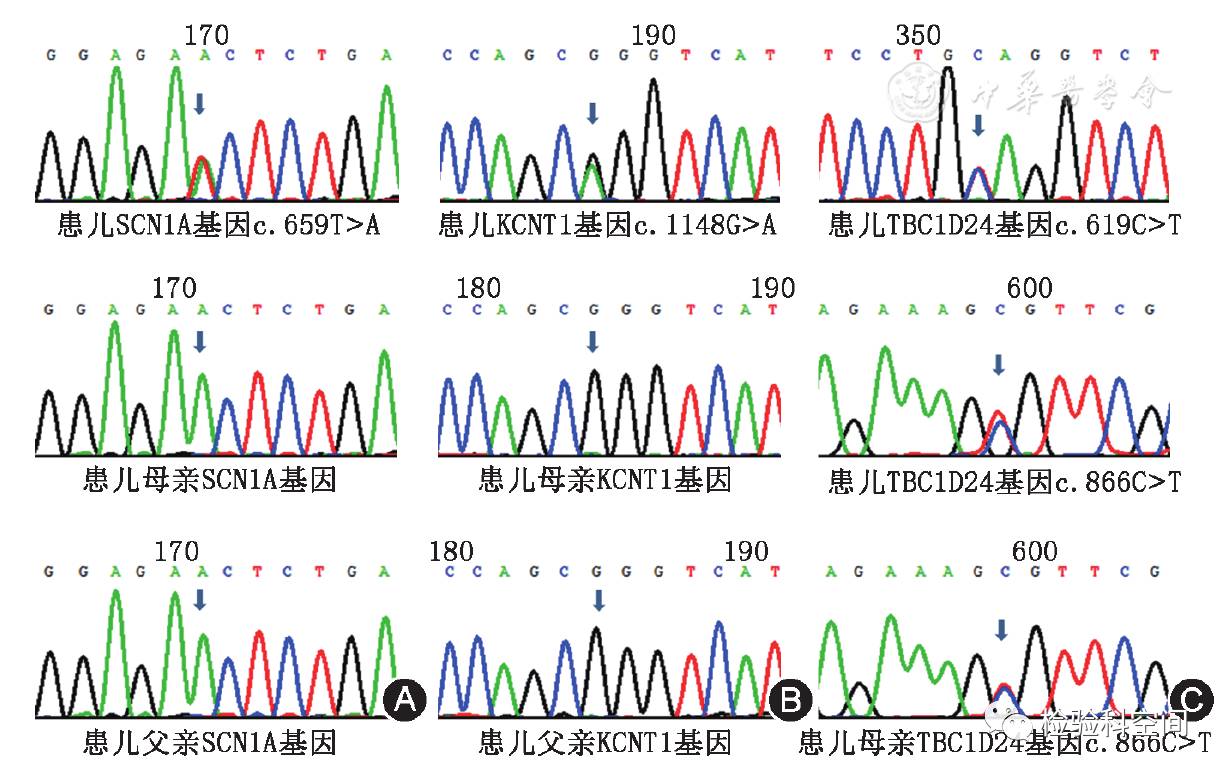
图2
婴儿癫痫伴游走性局灶性发作患儿及其父母基因测序图 A:例1患儿SCN1A基因c.659T>A突变,患儿父、母均无该位点突变;B:例5患儿KCNT1基因c.1148G>A突变,患儿父、母均无该位点突变;C:例7患儿携带TBC1D24基因突变c.866C>T和c.619C>T,患儿母亲携带TBC1D24基因突变c.866C>T
四、 治疗和随访
2例患儿失访,7例患儿随访2个月至5年8个月,均使用过3种或3种以上的抗癫痫药治疗,发作均未完全控制。其中2例服用氨己烯酸后最长发作控制时间为2周和2个月;2例服用奎尼丁,1例无效,1例最长发作控制1周。2例患儿尝试生酮饮食治疗,发作无明显减少。随访患儿中4例死亡(表1)。例1为SCN1A基因突变,该患儿曾使用丙戊酸、左乙拉西坦、托吡酯、苯巴比妥、奥卡西平、硝西泮、伊来西胺、氨己烯酸、扑米酮、唑尼沙胺并行生酮饮食治疗,发作未控制,28月龄死于癫痫持续状态。例4为KCNT1基因突变,曾使用鲁米那、托吡酯、硝西泮、左乙拉西坦、丙戊酸和促肾上腺皮质激素,发作未控制,68月龄死于癫痫持续状态。例2、3未发现致病基因突变,1例曾服用丙戊酸、左乙拉西坦、托吡酯和苯巴比妥,疑死于癫痫性猝死,另1例曾使用硝西泮、苯巴比妥和生酮饮食,死于癫痫持续状态。3例存活患儿智力、运动发育明显落后,分别随访8个月、1年及1年3个月仍不能竖头,不能翻身,不能逗笑。
讨论
EIMFS属于婴儿早期少见的难治性癫痫,国内外有关本病的报道较少,发病率不详。EIMFS患儿多在生后6个月内起病,生后40 d至3个月为高峰期,临床表现为游走性局灶性发作的特点。脑电图在发作期表现为游走性、多灶性放电,在一侧半球内或双侧半球之间游走,累及多个部位,临床发作与脑电图放电在时间和部位上密切相关[11]。患儿智力、运动发育落后。对抗癫痫药治疗反应不佳,通常预后较差,死亡率高。
Coppola[10]总结的EIMFS的自然病程进展大致可分为3个阶段:(1)第一阶段:通常起始于生后6个月内,表现为生后数周或数月,甚至生后第1天内出现零星的癫痫发作。可表现为局灶性发作伴泛化,可出现自主神经症状,如呼吸暂停、面色潮红或发绀。该阶段的脑电图在发作间期可表现为弥漫性背景活动减慢,以及由一侧大脑半球游走到另一侧的广泛性慢波。(2)第二阶段:年龄从1月龄至1岁不等,局灶性发作表现形式多样,且极其频繁,一日成簇发作5~30次不等,甚至连续几日近乎持续发作易形成癫痫持续状态。临床表现包括头眼向一侧偏转、眼睑眨动、单个或一侧肢体阵挛或强直发作、面色潮红和(或)发绀、咀嚼吞咽动作以及继发性全面强直阵挛发作。脑电图上的局灶性放电通常为游走性,但也可表现为局部固定的放电,同时在同侧大脑半球的其他区域或对侧大脑半球出现新的放电。(3)第三阶段:该阶段的年龄跨度较大,1至5岁不等,甚至更大。该阶段一般无发作,也可偶尔出现成簇发作或癫痫持续状态,且通常由其他疾病诱发。本研究的9例患儿均于上述病程的第一、二阶段诊断,随着病情进展,患儿发作逐渐频繁,抗癫痫药物难以控制。仅2例患儿1岁后发作较前减少,可能为进入病程自然进展的第三阶段,其中1例患儿为KCNT1基因突变,服用氨己烯酸后发作曾控制2周,后又出现发作。另1例患儿为SCN1A基因突变,服用氨己烯酸后发作最长曾控制2个月,该患儿发作有热敏感的特点,体温升高可诱发发作。9例患儿中4例死亡,死亡患儿中1例末次随访年龄为5岁8月龄,携带KCNT1基因突变,发作并未随年龄增长而减少,临床上尚未观测到进入病程第三阶段,对多种抗癫痫药物无效。
目前报道的与EIMFS发病相关的基因共有7个,分别为KCNT1[3]、SCN1A[4]、SCN2A[5]、SCN8A[6]、PLCB1[7]、SLC25A22[8]、TBC1D24[9]基因突变。部分患儿满足EIMFS的临床诊断标准,但未发现以上基因突变,仍可诊断为EIMFS。文献还报道过1例16号染色体片段重复(16p11.2)导致EIMFS的病例[12],重复的染色体片段包含24个已知基因,但尚未发现任何癫痫患者携带以上基因突变;在这24个基因中,推测3个基因(QPRT、DOC2A和SEZ6L2)突变可能影响癫痫发病。本研究的9例患儿中,2例SCN1A基因突变患儿均观测到热敏感现象,但不如Dravet综合征的热敏感特点突出。KCNT1基因突变在EIMFS中检出率最高,本组有3例。KCNT1基因突变除见于EIMFS外,还可见于大田原综合征等其他早发癫痫性脑病及常染色体显性遗传的夜间额叶癫痫[13]。文献报道1例TBC1D24基因突变家系,遗传方式为隐性突变,本研究中该基因突变的患儿携带来自母亲的错义突变(c.866C>T),并携带无义突变(c.619C>T),由于未取得患儿父亲的血样,尚无法验证该无义突变是否来自父亲,但考虑到无义突变的致病性较大,且患儿有典型的家族史及临床表现,故考虑该患儿符合EIMFS诊断,且为TBC1D24基因突变导致。
EIMFS为难治性癫痫综合征,多数患儿对各种抗癫痫药物的疗效欠佳。对于KCNT1基因突变的患儿,奎尼丁可显著抑制钾离子通道功能,从而控制发作[14]。目前国际报道奎尼丁治疗KCNT1基因突变导致的癫痫共3例[14,15],2例为EIMFS患儿,均对奎尼丁治疗有效,但发作均未控制,1例为常染色体显性遗传的夜间额叶癫痫,对奎尼丁无明显疗效。文献报道的奎尼丁用于治疗除疟疾以外儿科疾病的用量[15]为15~60 mg/(kg·d),每日最大剂量为3 000~4 000 mg,奎尼丁用于KCNT1基因突变靶向治疗的药物剂量尚无明确标准。本组2例KCNT1基因突变的患儿服用奎尼丁后发作均有减少,但发作未控制,且由于奎尼丁对心脏的不良反应,如QT间期延长,限制了其调整药物剂量的空间。目前国际上对奎尼丁治疗KCNT1基因突变导致的癫痫尚未达成共识。对于其他基因突变的病例,尚无相关靶向药物的报道。国外有关于溴化钾[16]、乙酰唑胺[17]、卢非酰胺[18]、生酮饮食[19]等对EIMFS有疗效的报道,本组2例尝试生酮饮食治疗无效。
参考文献:略