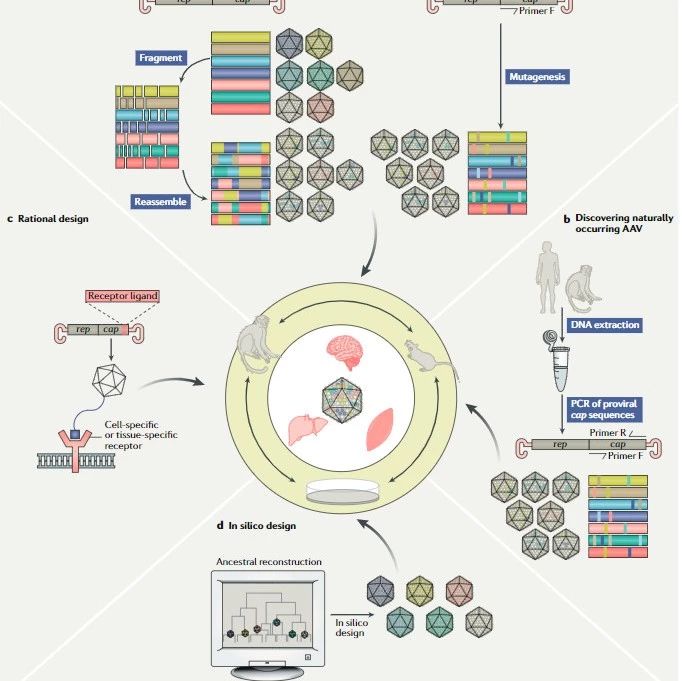
渐冻人与基因检测
前言


肌萎缩侧索硬化(ALS)是一种成人发病的神经系统退化性疾病,俗称“渐冻人症”。其特点为上、下运动神经元缺失,渐进性的麻痹,在症状出现后平均2–5年后死亡。诊断是基于临床特点、电学检查和排除其他疾病重叠的症状。姑息性治疗,目前除了利鲁唑片没有有效药物,利鲁唑片会减慢疾病进展,平均延长生存期3个月。大多数患者是散发性的ALS(sALS),大约5–10%的ALS患者有家族史 (家族性ALS(fALS)。临床上fALS和sALS相似。然而,fALS患者特点是发病的平均年龄(46岁)相比sALS (56岁)更早。fALS的病情进展会大大短于或长于sALS,这可能与特定的基因突变有关。虽然将ALS分类为家族或散发性对临床实践有用,但是fALS和sALS生物和遗传特点日益模糊是确定的。
ALS患者经常问他们为什么患上这种病,他们会把它传给孩子的机率是多少,他们的病情发展有多快。从统计数据看,大多数病人(sALS)可以放心,在他们的家系中没有其他人处于危险之中。fALS患者被告知他们可以遗传给子女,因为基因检测仅发现SOD1突变占fALS患者的20%,大多数患者不得不接受他们的致病基因无法明确的事实。近年来,对于ALS基因检测及遗传咨询已转移到发现新致病基因层面包括C9orf72,ALS和额颞部位痴呆(FTD)之间联系的认识和下一代测序技术的出现。目前2/3的fALS和10%的sALS的遗传病因可以明确。2011年发现基因C9orf72六核苷酸重复序列使大多数fALS和sALS致病性变异得到确认,揭示了ALS 和 FTD的遗传病因。虽然ALS患者渴望获得基因检测,但在ALS患者的临床护理方面,遗传学研究进展是缓慢的。
在这篇文章中,我们回顾了对ALS病人遗传学的现有理解,指出遗传咨询的注意事项,并总结了当ALS病人需要选择合适的基因检测时所需要考虑的检测项目及其影响因素。这些信息可能对于提供给ALS患者看护人和遗传学家,遗传谘询人员,神经病学家和其他的医生有所帮助。伴随着获得基因检测和遗传咨询的增加,ALS患者和家庭可从ALS遗传学领域的最新进展中受益。
ALS遗传学概述
通过对fALS家系的研究,已发现了大于25个 ALS致病基因,它占fALS患者的2/3,约占sALS患者的11% (如下图)。
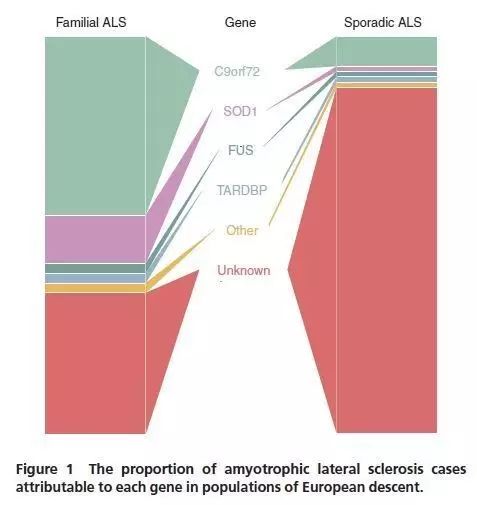
大多数ALS基因是以显性方式遗传,有变异和年龄相关的外显率。家系内部和家系间在发病年龄和疾病进展方面变化的重要性是可以观察到的。可能会导致隐性遗传的基因包括OPTN, SPG11, FUS以及 SOD1(明确的,Asp90Ala纯合突变),还有UBQLN2是X连锁显性基因。在这里,我们回顾最常见的ALS基因C9orf72, 第一内含子六核苷酸(G4C2)重复序列,它占了大约fALS患者的40%、sALS 患者的7%、家族性FTD患者的25%和多达88%ALS合并FTD的患者。这些频率远远超过任何其他ALS或FTD的基因;然而致病C9orf72的重复序列频率在种族/地理来源方面相差很大。据报道,在欧洲北部(特别是北欧)频率最高,在亚洲频率低。到目前为止,C9orf72重复序列变异是唯一在sALS患者中发现的频率大于2%的ALS突变。
C9orf72
C9orf72变异位点GGGGCC(六核苷酸)重复次数范围从2到4000多。重复次数小于25通常被认为是正常的。重复多少为致病的阈值并不完全清楚。正如Gijselnick et al所述,在FTD和ALS患者中,由于可变的实验室检测方法和不确切的规模较大的重复,建立一个明确的导致致病性的重复次数的阈值是复杂的。此外,对于重复的携带者个体内重复大小的变异可能是重要的。与非神经组织相比,更大的重复发生在神经系统。虽然有预测的文献报道,但这并没有被明确的规定为ALS/FTD C9orf72的一个特征。
C9orf72重复序列变异是以常染色体显性的方式传递,具有可变性和年龄相关的外显率。大多数受影响的个人的父母亲也受影响。然而,缺乏家族史,缺乏对父母的疾病的识别,在症状出现之前,父母就去世了以及其他情况会导致一个阴性的家族史。早期的研究已经估计了年龄相关的外显率在35岁为0%,在58岁为50%, 在80年将近100%。要了解C9orf72重复序列的外显率需要进一步的研究。临床目前正在努力研究使用寡核苷酸(ASOs) 等位基因特异性来治疗C9orf72重复变异的病理表现.
SOD1
SOD1是第一个通过连锁分析被确认为fALS病因的基因。SOD1突变是在C9orf72之后引起fALS的第二个最常见的已知基因。据报道他们发生在大约12%的fALS,大约1–2%的sALS中。SOD1突变的频率差别很大,在伊朗发生在38%的fALS,在爱尔兰0%。临床上SOD1突变通常是与认知功能障碍不相关,与其他fALS类型相比延髓发病是不太常见。一些表现型与基因型的关连情况是已知的;在北美Ala5Val突变(指老年报告Ala4Val)占SOD1突变的一半,发展迅速,症状出现1年内死亡。其他突变与一个长期10年或更长的病程相关,包括Gly37Arg,Gly41Asp,His46Arg,Glu100Lys,4,6和Asp90Ala。SOD1突变通常是以常染色体的显性方式遗传。然而,在北部斯堪的纳维亚半岛Asp90Ala突变以隐性方式遗传。在其他人群可能占主导地位。显性的SOD1突变与易变的年龄相关的外显率有关,外显数据可能高估了风险,由于高外显突变家系偏倚的确认。在一个大的谱系Ile113Thr突变外显率估计为50 %(60岁)和80%(88岁)。针对SOD1寡核苷酸治疗的第一期临床试验在2012年完成,与安慰剂相比并没有副作用。一些针对SOD1突变携带者的临床试验正在研究治疗方法。
TARDBP
TARDBP突变是显性遗传,发现在大约4%的fALS和1%sALS。fALS的这一突变基因发现之前,其蛋白产物(TDP-43)被认为是ALS and FTD的泛素阳性神经元包裹体的成分。TARDBP突变已经在不同地域的ALS家系中被确定,在撒丁岛高频率Ala382Thr突变起到奠基者效应。TARDBP相关ALS被认为有典型的临床表现,可能包括FTD和少见的没有运动神经元疾病的FTD。
FUS
据报道FUS突变约占fALS的4%和大约sALS的1%,以最高的频率(8.7%)确定出现在德国fALS患者中。在多个家系中确定的经常发生的突变包括Arg521Cys 和 Arg521Gly。回顾了20个受影响的个人的临床信息显示性别比例,平均发病年龄44.5岁,颈椎发病10个人、腰椎发病五人,延髓发病三人。FUS突变在FTD患者中已有报导。FUS突变与寿命长短有关,虽然对广泛的家族内部的变化进行了观察。截短突变在早发性和侵略性病程患者中已被确认,最近新生FUS突变被证明是德国早发性ALS(发病年龄小于35岁)患者最常见的病因。FUS突变通常是显性遗传,尽管His517Gln纯合的隐性突变已被报导在佛得角血统的fALS家系中。
遗传咨询和基因检测
与所有临床遗传评估一样,家族史是ALS遗传风险评估及遗传咨询的一个基本组成部分。临床医生应该记录一个三代家系(至少)确定ALS,FTD,其他痴呆症,帕金森病、精神障碍和自杀。家系图应被用来回顾显性遗传的证据,因为大多数ALS基因是显性遗传。然而外显不全、不完整的家系信息、误诊,错位,早死,未公开的收养和其他情况可能会掩盖真实信息。例如,受影响的兄弟姐妹与经典的隐性遗传相关联,但在ALS患者中可以看到以显性方式遗传伴随非完全外显性,早死,或隐瞒父母的遗传状态的其他问题。
在ALS患者一级或二级亲属没有ALS或FTD的家族史,可以根据经验数据估算亲属发展为ALS的可能性。一级亲属的ALS的终生风险为1–3%(男性亲属3%,女性亲属范1%)。二级亲属的风险更低,更多的远亲家系成员的风险似乎没有增加。没有密切家族史的ALS或FTD的欧洲血统的先证者更适合讨论这个风险信息。关于ALS在其他人群中的家族性聚类数据是不容易得到的。C9orf72重复序列的不同的地理分布是欧洲经验数据不适用于非欧洲血统人的原因之一。
对一级或二级亲属有ALS或FTD的ALS患者,家系成员的遗传风险评估应告知家人,通过家系分析和/或基因检测。如果是显性遗传,那么子女有50%的机会遗传一个ALS突变。据估计,高外显突变的外显率估计为60–70%。同时发生ALS和FTD的患者大多数有阳性家族史是由C9orf72重复序列变异引起。
基因检测方案
目前ALS基因检测包括Sanger测序,分析C9orf72重复序列次数,多基因检测,下一代测序panel(这是与C9orf72重复序列次数检测的黄金搭档)和全基因组测序。目前可用的检测,预计的周转时间和成本,表1中列出。
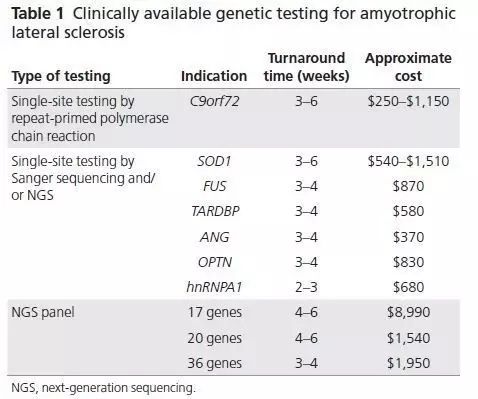
此表中显示的ALS基因检测是是在起草本文时,由大的商业实验室提供,有关最新的信息,在检测时应咨询网站或特定的实验室工作人员.
有几个因素可以考虑作为所有欧洲或部分欧洲血统的ALS患者C9orf72重复序列检测的原因,不管家族史怎样。在sALS C9orf72重复序列的频率为7%,检测的成本相对较低,检测结果有潜在的重要性,当前和即将来临的以C9orf72为目标的临床治疗试验可能会证明这一做法的正确性。对于C9orf72检测为阴性、非欧洲的fALS患者应当提供多基因检测(请参见图2)。
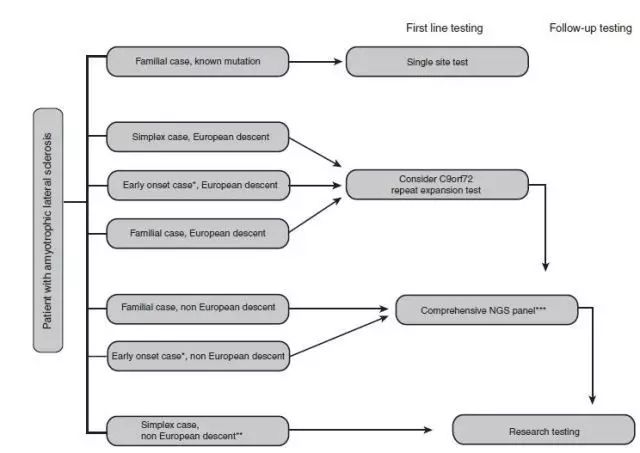
基于我们现有的临床经验,此方法在fALS患者确定的致病突变为63.6%,与发表的文献相符。这种方法潜在的缺点是不能鉴定第二个有害突变或C9orf72呈阳性的患者的变异。家系中有多个分离的ALS突变或者变异已被确定,比预期的频率更大。在这种情况下C9orf72的重复检测是阳性的,作为第一步,确认每一个受影响家系成员的重复序列尤为重要。
此外,还应考虑提供检测的sALS患者早期发作的症状(50岁以下),虽然在这些人中基因检测的大量的数据还不能获得。有限的证据表明,有阴性的家族史的早发性ALS患者可能有新生突变。如果早期发作患者的C9orf72检测是阴性的,那么多基因panel检测可被建议。尽管全基因组测序在确定ALS基因研究方面富有成效,但ALS患者全基因组测序的临床评估是未知的。在fALS家系通常只有一个受影响的家系成员存活并进行检测,从而限制了家系分离分析的机会。如果C9orf72重复序列检测是阴性的在某些情况下可以考虑外显子组测序。早期发病和父母可用于检测/变量筛选的sALS先证者是适当的外显子组测序的候选人。fALS患者有两个或更多受影响的人可供检测的人可以考虑作为外显子组测序的候选人。
测试前咨询
测试前遗传咨询能帮助患者预测自己及他们的家系成员基因检测的可能影响。认知障碍的病人应由合法监护人陪同或医疗机构代理。应强调ALS基因的异质性和可变外显率。基因检测的局限性应告知患者和家属,包括:(i)阴性的结果的ALS先证者并不能排除遗传基础或贡献ii)如果一个不确定意义的变异被发现,那么该检测是不明确的。(iii)阳性结果与病程预测不会总是一致。不愿意接受基因检测的家庭可以考虑基因库作为未来的检测。
症状前基因检测
症状前基因检测可以给ALS患者的成人一级亲属确定突变。有风险家系成员可能有许多理由寻找症状前基因检测:为了减少不确定性,为将来打算,为了健康和生活方式的选择,和有关计划生育的决定。在过去,ALS的症状前基因检测的指导方针在亨廷顿舞蹈病和阿尔茨海默病的协议之后已经被定制其中包括检测前的遗传咨询,神经系统疾病基线和认知评估、心理评估,披露的人,支持的人,和测试后的遗传咨询。最近,Benatar et al研究了ALS患者症状前基因检测并为实践提出了详细建议。这些作者建议测试过程中应至少包括两个遗传咨询环节(其中可能包括检测前和检测后咨询),特定的电话咨询是可以接受的。建议至少间隔1周的时间在初步遗传咨询和做出接受测试的决定之间,以便有足够时间接受个人信息和作出明智的决定。
检测后的咨询
个人收到阳性检测结果,应当被告知这些信息:关于基因,具体的突变,已确立的基因型-表型关系。在突变的遗传模式的背景下应回顾家族史,家系成员包括后代的复杂关系和疾病风险应当被回顾。应该讨论不完全外显的可能性。fALS家系往往提出疾病为什么会发生,这些应该被探讨和解决处理。临床医生应为家系成员提供一个基因检测结果的副本,促进亲属检测的准确性正如一个简要的总结概述。这些信件通常作为通知基因诊断的家系成员的载体。这在ALS患者中尤为重要因为先证者可能会死或在收到结果之前不能亲自来为沟通。
总结
当前ALS管理指南没有解决基因检测的问题,最近发现的新基因和技术方面的进步是复杂的同时又优化了遗传风险评估的过程,基因检测和ALS的咨询。不久前,ALS基因检测仅限于SOD1测序;临床医生现在有各种各样的检测方法可以选择,包括C9orf72重复次数检测,多基因NGS, 全外显子组测序。然而基本的临床技能,如家族史、系谱分析和风险评估对于基因检测和咨询的合理应用仍然是至关重要的。基因检测可能会帮助ALS患者了解他们的情况的基础上给出准确的风险评估和家系成员的遗传检测以及特定的基因型的治疗方案。正如fALS和sALS的部分的遗传致病机理已被阐明,基因检测和咨询将会变得越来越重要,应当纳入到ALS的常规管理中。
本文翻译自
Genetic testing and genetic counseling for amyotrophic lateral sclerosis: an update for clinicians.
Genet Med. 2017 Mar;19(3):267-274. (PMID:27537704,回复数字获得全文下载链接)
译者介绍

孙海瑞,男,医学硕士,从事单基因遗传病基因测序数据解读工作3年余,现就职于智因东方转化医学研究中心有限公司,任临床组组长