内源基因激活表达系统dCas9-SAM,以及操作手册和详细慢病毒包装手册
dCas9-SAM: an engine for activation the endogenous gene expression
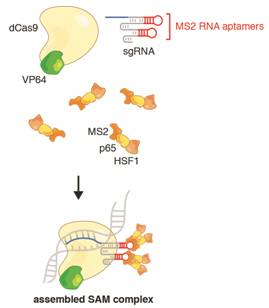
Synergistic activation mediator (SAM) is developed at the basis of CRISPR/Cas9 technique. Wildtype cas9 has been mutated at amino acid positions D10A and H840A which completely inactivate both the nuclease and nickase activities. The resulted double mutated cas9 is called as dcas9. With the help of a target-specific guide RNA (sgRNA), it can bind to a specific region of DNA without cutting it. In this context, dCas9 carries a C-terminal tripartite synergistic activation mediator (SAM). By targeting dCas9-SAM to a promoter region, we can specifically induce expression of virtually any gene of interest.
Plasmids for this protocol
The SAM system involves three lentivirus plamids. Lenti sgRNA(MS2)_puro have BsmBI restriction enzyme site, which is for insertion of the oligo sgRNA under the hU6 promoter. The result sgRNA(MS2) lentivirus express the fused MS2 and specific sgRNA while gives the cells the puromyucin resistance. Lenti dCas9-Vp64 has fused dcas9 and vp64 cassette drived by EF1α promoter and has the Blastcidin resistance. The third plamids for SAM system is lenti MS2-p65-HSF1, while it has a distinct hyromucin resistance genes. To sum up, SAM system is widely used for the activation of interest gene by targeting the activation regulatory factors to the promoter the region. It needs to packet three lentivirus harboring the all components of the system with distinct resistance genes.

Procedure:
Ø 1.Design the sgRNA_SAM at the follwing website
Optimized sgRNAs for any coding gene can be found using SAM Cas9
activator design tool designed by ZhangLab, MIT at the following website:
http://sam.genome-engineering.org/database/
Synthesize two oligos of the following form.

Example for sgRNA oligo design: Note that the NGG PAM is not included in the designed oligos.

Tips: Standard de-salted oligos (usually the most inexpensive
synthesis) are sufficient for cloning. If not already resuspended, dilute each oligo to 100
μM in sterile water or TE.
Ø 2. Insert the SgRNA oligo into lenti sgRNA(MS2)_puro plasmid, the map of lenti sgRNA(MS2)_puro is as follows:

1. Oligo annealing
|
Oligo-Forward (100 μM) |
1 μl |
|
Oligo-Reverse (100 μM) |
1 μl |
|
10×T4 ligase buffer (NEB) |
1 μl |
|
T4 PNK(NEB) |
1 μl |
|
ddH2O |
6 μl |
|
Total volume |
10 μl |
Mix the components above and anneal in a thermal cycler with the following conditions:
On thermal cycler with heat lid.
37 ℃, 30 min
95 ℃, 5 min
Ramp down to 25℃ at 0.1℃/sec. (when step 25℃ is highlighted, click option, set Ramp as 0.1℃/sec)
25 ℃, 5 min
4 ℃, 5 min
Put on ice, or keep in -20℃ freezer for storage.
2. Digest lenti sgRNA(MS2)_puro plasmid with BsmBI
|
Plasmid |
3 μg |
|
BsmBI |
2 μl |
|
10 × buffer |
5 μl |
|
ddH2O |
Up to 50 ul |
|
Total volume |
50 ul |
Tips: Purify the enzyme digested product with PCR product purification kit, and make sure that the concentration is more than 50 ng/ul.
3. Ligation
Dilute the annealed target oligos 200 times for further ligation
|
Diluted annealed oligos |
2 μl |
|
Digested plasmid |
50-100 ng |
|
10× T4 ligase buffer |
1 μl |
|
T4 ligase |
1 μl |
|
ddH2O |
Up to 10 ul |
|
Total volume |
10 ul |
Ligate for more than 2 hours at 22℃. (Overnight is better.)
4. Transformation and identification of positive clones
Transform the ligation product into the stabl3 competence cells and pick single clones and culture for several hours in 37℃ incubator, and perform PCR to identify the positive clones.
|
sgRNA(MS2)-F |
GAG GGC CTA TTT CCC ATG ATT CCT TCA TAT |
Use Oligo-reverse and sgRNA(MS2)-F as primers, and the PCR product is about 350 bps.
Note: sgRNA(MS2)-F is the sequencing primer for oligo sgRNA
5. Plasmid extraction and sequencing validation
Culture the positive clone in 10 ml Amp+ LB medium, and extract plasmid. UsesgRNA(MS2)-F as sequencing primer.
6. Lentivirus production
Lentiviral Production Protocol
The day before transfection:
The day before transfection, seed around 5×106 HEK293FT cells per 100mm petri dishes.
The day of transfection:
1. Check to make sure cells are at least 70% confluent before transfection
2. Combine the appropriate amount of plasmid DNAs in a 1.5ml conical tube:
For per100mm petri dish:
10 μg Lentivirus backbone plasmid harboring inteterest gene
4 μg pdR8.2. (structural vector)
2 μg pMDG2 (envelope vector)
16 μg in total
3. Add filtered water to a final volume of 878μ1.
4. Add 122μ1 2M CaCl2 and mix.
5. Drop 1000ul of 2xHBS to the abvove mixture slowly. Note: Shaking the tube continuously to avoid generating large particles
6. Sit mixture at room temperature for 2min.
7. Remove old media and replace with 9 ml new complete culture media.
8. Add all of mixture to the cells gently and evenly. Swirl the petri dish to distribute evenly and put it back to incubator.
9. 8 hours later, Change media to remove the transfection reagent and replace with 12 ml new complete culture media
10. Generally, 72 hours after transfection , harvest cell culture supernatant containing lentivirus and filter through a 0.45μm filter.
11. Concentrate the lentivirus with PEG8000 or with ultra-speed centrifugation.
Transfection reagent stock preparation:
2 M CaCl2
2xHBS:50mM HEPES
10mM KCl
12mM Dextrose
280mM NaCl
1.5mM Na2HPO4
Resuspend in 900 ml H2O, adjust pH to 7.04 (with NaOH if using HEPES free acid, with HCl if using HEPES sodium salt). Raise volume to 1000 ml and carefully re-adjust final pH to 7.05 exactly (Note: pH needs to be very precise at pH 7.04 – 7.05). Filter through 0.22μM and store in aliquots at -20°C (good for up to 6 months).
Suggestions and Tips: You can also choose other transfection reagents including lipofectamine2000, PEI and Turbofect (Thermo/Fermentas). Remember that the transfection efficiency should be over 90 percent.